forked from natverse/hemibrainr
-
Notifications
You must be signed in to change notification settings - Fork 0
Commit
This commit does not belong to any branch on this repository, and may belong to a fork outside of the repository.
- Loading branch information
1 parent
77da93c
commit cd57920
Showing
78 changed files
with
14,738 additions
and
7 deletions.
There are no files selected for viewing
This file contains bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
| Original file line number | Diff line number | Diff line change |
|---|---|---|
|
|
@@ -7,10 +7,5 @@ packrat/lib*/ | |
| examples | ||
| .httr-oauth | ||
| .Renviron | ||
| inst/doc | ||
| docs/ | ||
| *hemibrain_data/ | ||
| data-raw/annotations/ | ||
| doc | ||
| Meta | ||
| docs | ||
Binary file not shown.
This file contains bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
This file contains bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
This file contains bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
| Original file line number | Diff line number | Diff line change |
|---|---|---|
| @@ -0,0 +1,42 @@ | ||
| ## ----setup, include = FALSE--------------------------------------------------- | ||
| knitr::opts_chunk$set( | ||
| collapse = TRUE, | ||
| comment = "#>" | ||
| ) | ||
|
|
||
| ## ----save.matches, eval = FALSE----------------------------------------------- | ||
| # # Load package | ||
| # library(hemibrainr) | ||
| # # See matches | ||
| # View(hemibrain_matched) | ||
|
|
||
| ## ----fresh.matches, eval = FALSE---------------------------------------------- | ||
| # matches = hemibrain_matches() # You will be asked to log-in through a Google-enabled email address. | ||
| # View(matches()) | ||
|
|
||
| ## ----ready.made, eval = FALSE------------------------------------------------- | ||
| # made.matches = read.csv("my_matches.csv") # Must have the named columns: bodyid, skid, quality | ||
| # hemibrain_matches(df = made.matches, direction = "both") # direction controls which tabs matches get written to | ||
|
|
||
| ## ----add, eval = FALSE-------------------------------------------------------- | ||
| # # Add a mising FAFB projection neuron, so we can match it later: | ||
| # hemibrain_matching_add(ids = "16", sheet = "fafb", User = "ASB") | ||
| # ## the sheet argument specifies the worksheet or 'tab' on the Google sheet we want to add to | ||
|
|
||
| ## ----interactive.pipeline, eval = FALSE--------------------------------------- | ||
| # # install package to bridge neurons between FAFB14 and hemibrain space | ||
| # if (!requireNamespace("remotes")) install.packages("remotes") | ||
| # remotes::install_github('natverse/nat.jrcbrains') | ||
| # nat.jrcbrains::download_saalfeldlab_registrations() | ||
| # | ||
| # # Match hemibrain neurons! | ||
| # hemibrain_matching() # Automatically, you can choose a User ID and you are given neurons that have this ID in the User column on the Google Sheet. | ||
| # hemibrain_matching(ids=c("674108632","739256609")) # Otherwise you can select specific IDs | ||
| # hemibrain_matching(ids=c("674108632","739256609"), overwrite = TRUE) # If a match has already been made you can overwrite it | ||
| # # Otherwise neurons that have already been given a match will not be shown in the pipeline. | ||
| # | ||
| # # Match FAFB neurons! | ||
| # fafb_matching() | ||
| # fafb_matching(ids = "16") # Specify IDs | ||
| # fafb_matching(ids = "16", overwrite = TRUE) # Overwrite | ||
|
|
This file contains bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
| Original file line number | Diff line number | Diff line change |
|---|---|---|
| @@ -0,0 +1,176 @@ | ||
| --- | ||
| title: "match_making" | ||
| output: rmarkdown::html_vignette | ||
| vignette: > | ||
| %\VignetteIndexEntry{match_making} | ||
| %\VignetteEngine{knitr::rmarkdown} | ||
| %\VignetteEncoding{UTF-8} | ||
| --- | ||
|
|
||
|
|
||
| ```{r setup, include = FALSE} | ||
| knitr::opts_chunk$set( | ||
| collapse = TRUE, | ||
| comment = "#>" | ||
| ) | ||
| ``` | ||
|
|
||
| > "Oh, but nonsense, she thought; William must marry Lily. They have so many things in common. Lily is so fond of flowers. They are both cold and aloof and rather self-sufficing. She must arrange for them to take a long walk together." | ||
| ([Virginia Woolf](https://en.wikipedia.org/wiki/To_the_Lighthouse)) | ||
|
|
||
| # Neuron Matching Making | ||
|
|
||
| Insect brains seem pretty stereotyped. But just how stereotyped are they? It comes as a suprise to many neuroscientists who work only on vertebrates, to learn that in insects, individual neurons can readily and reliably be re-found and identified across different members of the species. Perhaps even across species. | ||
|
|
||
|  | ||
|
|
||
| As of 2020, two large data sets for the vinegar fly, D. melanogaster, are available making it possible to look at the full morphology of ~25,000 neurons in two data sets. These data sets are the [hemibrain](https://neuprint.janelia.org/help/videos?dataset=hemibrain%3Av1.0&qt=findneurons&qr%5B0%5D%5Bcode%5D=fn&qr%5B0%5D%5Bds%5D=hemibrain%3Av1.0&qr%5B0%5D%5Bpm%5D%5Bdataset%5D=hemibrain%3Av1.0&qr%5B0%5D%5Bpm%5D%5Binput_ROIs%5D%5B0%5D=ATL%28R%29&qr%5B0%5D%5Bpm%5D%5Boutput_ROIs%5D%5B0%5D=IB&qr%5B0%5D%5BvisProps%5D%5BrowsPerPage%5D=25&tab=0&q=1) and [FAFB](https://neuropil.janelia.org/tracing/fafb/v14/). However, neurons in FAFB have been semi-manually or manually reconstructed, making the automatic assignment of FAFB-hemibrain neuron matches non-trivial. In this R package we have built tools to enable users to record and deploy inter-dataset matches. | ||
|
|
||
| What use is this information? Matches could be used to look at morphological stereotypy, help find genetic lines that label neurons, help transer information associated with on reconstructed to the same cell in a different brain, compare neuron connectivity between two brains, etc. | ||
|
|
||
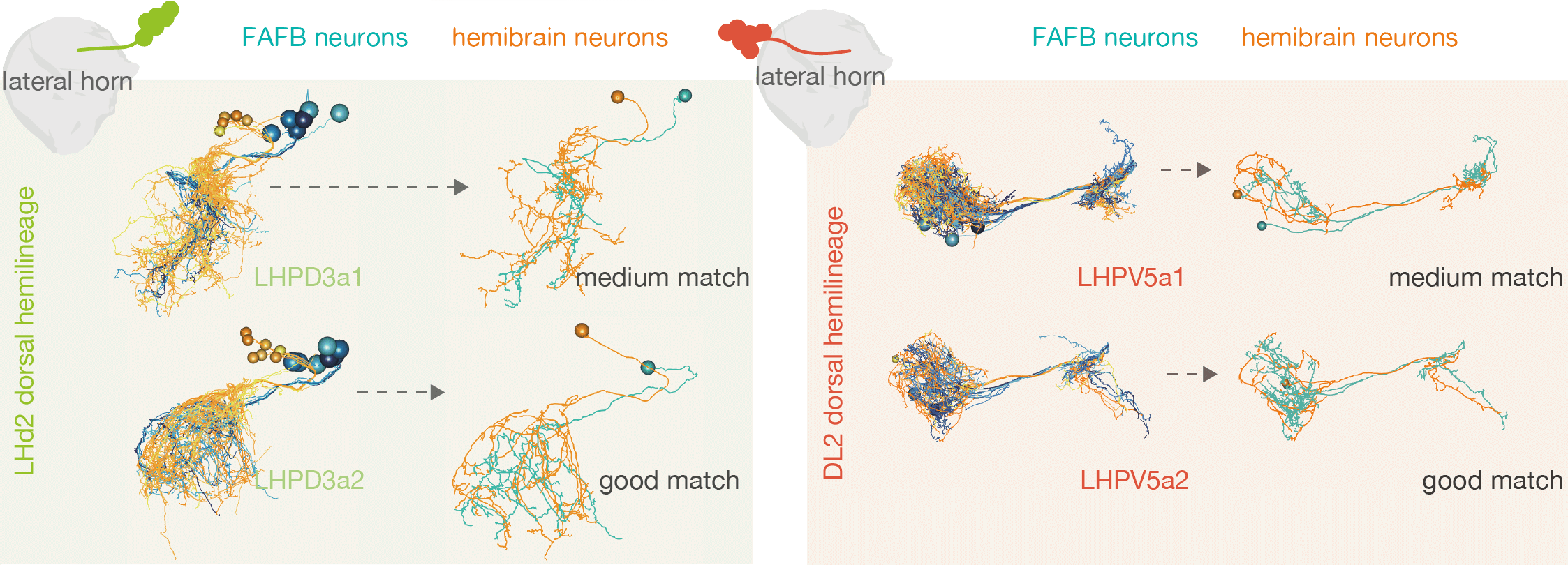
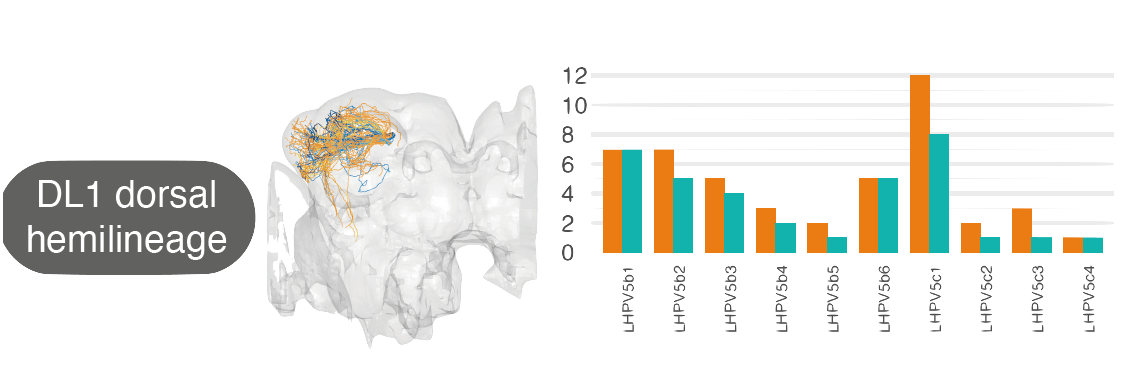
| For example, by matching neurons up between the hemibrain and FAFB, we see that the numbers of [cell types](https://pubmed.ncbi.nlm.nih.gov/30703584/) within one 'hemilineage' (se set of neurons that are born and develop together) are comparable between these two different flies: | ||
|
|
||
|  | ||
|
|
||
| ## What You need | ||
|
|
||
| In order to use these tools you will need to have [RStudio](https://rstudio.com/) and to have installed the [natverse](http://natverse.org/). To use them to maximum effect, you will need to also have permission to access the [FAFB CATMAID v14 project](https://neuropil.janelia.org/tracing/fafb/v14/), although some neurons are available to be read by the public from [Virtual Fly Brain](https://v2.virtualflybrain.org/org.geppetto.frontend/geppetto)'s CATMAID project for FAFB and should have the same unique skeleton ID numbers. Our pipeline function makes use of the [Google Filestream](https://support.google.com/a/answer/7491144?hl=en) application, which should be installed on your machine. Further, note that neurons are read from the FAFB CATMAID project, and you must have login details for this project recorded in your .Renviron (edit with: usethis::edit_r_environ) for these functions to work. For help, see [here](https://github.com/natverse/rcatmaid). | ||
|
|
||
| ## Authorisation | ||
|
|
||
| In order to write neuron matches to the project you must have access to the `hemibrain` Google Drive or the match making Google sheet (see below) owned by the [Drosophila Connectomics Group](https://www.zoo.cam.ac.uk/research/cell-and-developmental-biology/drosophila-connectomics#:~:text=The%20project%20aims%20to%20reconstruct,Research%20Campus%20in%20the%20US.). If you do not have access but would like to help or use this information, get in contact! You do not need programming skills to to help us match make neurons, as we have written an interactive pipeline in R which does most of the work for you (see below). | ||
|
|
||
| We regularly also up-date a data frame saved in this package, as a snapshot of matches that have been made. Without authorisation you can access these matches but they may not be the most up-to-date: | ||
|
|
||
| ```{r save.matches, eval = FALSE} | ||
| # Load package | ||
| library(hemibrainr) | ||
| # See matches | ||
| View(hemibrain_matched) | ||
| ``` | ||
|
|
||
| ## The Google Sheet | ||
|
|
||
| We in the [Drosophila Connectomics Group](https://www.zoo.cam.ac.uk/research/cell-and-developmental-biology/drosophila-connectomics#:~:text=The%20project%20aims%20to%20reconstruct,Research%20Campus%20in%20the%20US.) have been recording our match making in a Google sheet named [em_matching](https://docs.google.com/spreadsheets/d/1OSlDtnR3B1LiB5cwI5x5Ql6LkZd8JOS5bBr-HTi0pOw/edit). This sheet has two tabs of concern here, `hemibrain` for hemibrain neuron -> FAFB neuron matches and `fafb` for FAFB neuron to hemibrain neuron matches. | ||
|
|
||
| 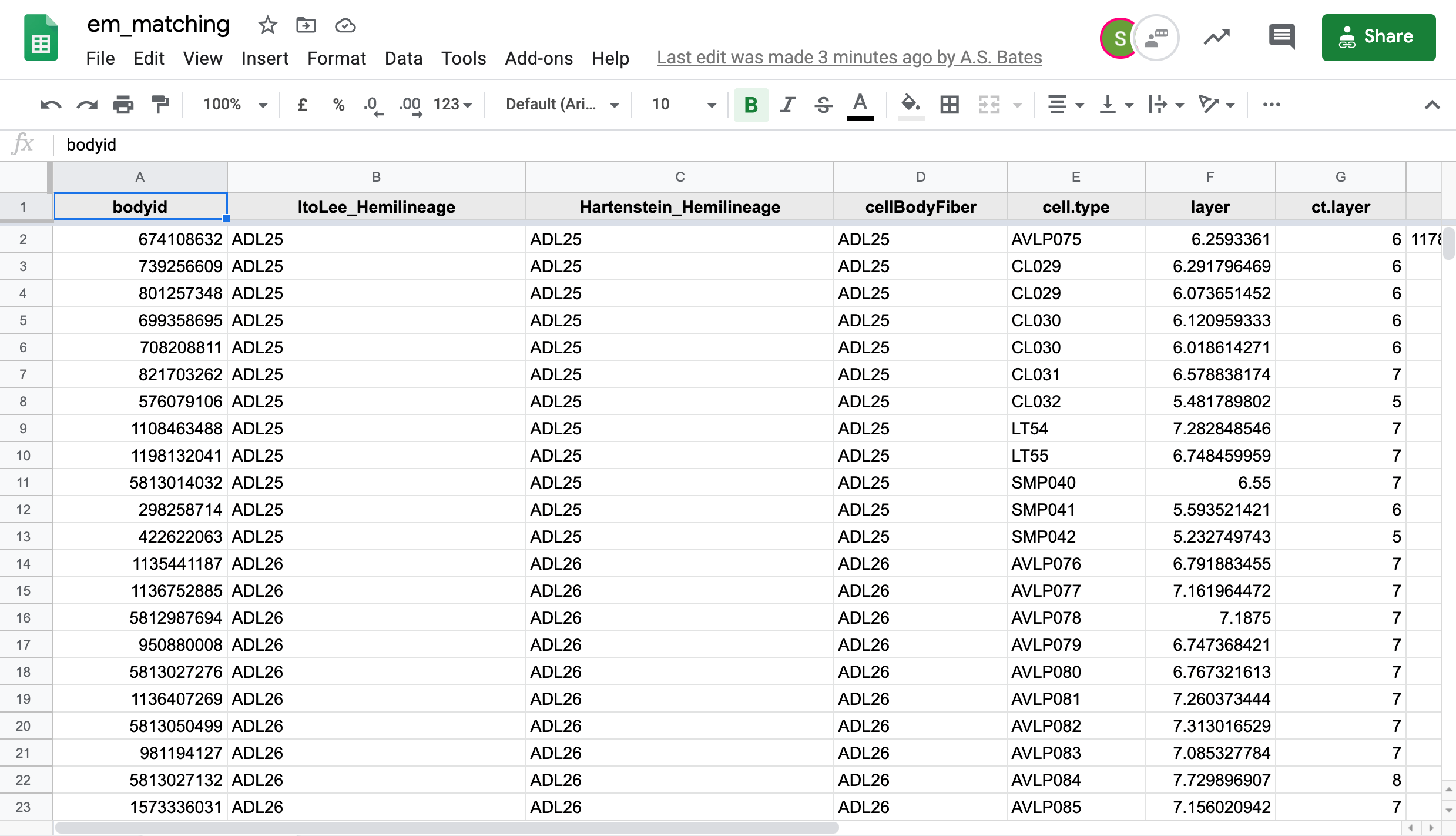 | ||
|
|
||
| If you have authorisation, you can see the most up-to-date matches as so: | ||
|
|
||
| ```{r fresh.matches, eval = FALSE} | ||
| matches = hemibrain_matches() # You will be asked to log-in through a Google-enabled email address. | ||
| View(matches()) | ||
| ``` | ||
|
|
||
| As you can see, other meta information is present in the data frame `matches`. The function `hemibrain_matches` has an argument called `priority`. This specifies whether to use FAFB->hemibrain matches (FAFB) or hemibrain->FAFB matches (hemibrain) in order to ascribe cell type names to FAFB neurons. In both cases, cell type names are attached to hemibrain bodyids, and propagated to their FAFB matches. | ||
|
|
||
| ## Match Quality | ||
|
|
||
| Once a match is recorded, the user selects a quality for that match. There can be no match (none), a tract-only match (tract) a poor match (poor) an okay match (medium) or an exact match (good). As a rule of thumb, a poor match could be a neuron from a very similar same cell type or a highly untraced neuron that may be the correct cell type. An okay match should be a neuron that looks to be from the same morphological cell type but there may be some discrepancies in its arbour. A good match is a | ||
| neuron that corresponds well between FAFB and the hemibrain data.A tract only match just means that the matched neuron should share the same cell body fiber, and therefore same developmental ontogeny, even if the rest of its morphology is quite different. | ||
|
|
||
| It is very important to note that a match cannot be a match if neurons do not seem to share the same cell body fiber tract. Being in a different tract is a deal breaker. | ||
|
|
||
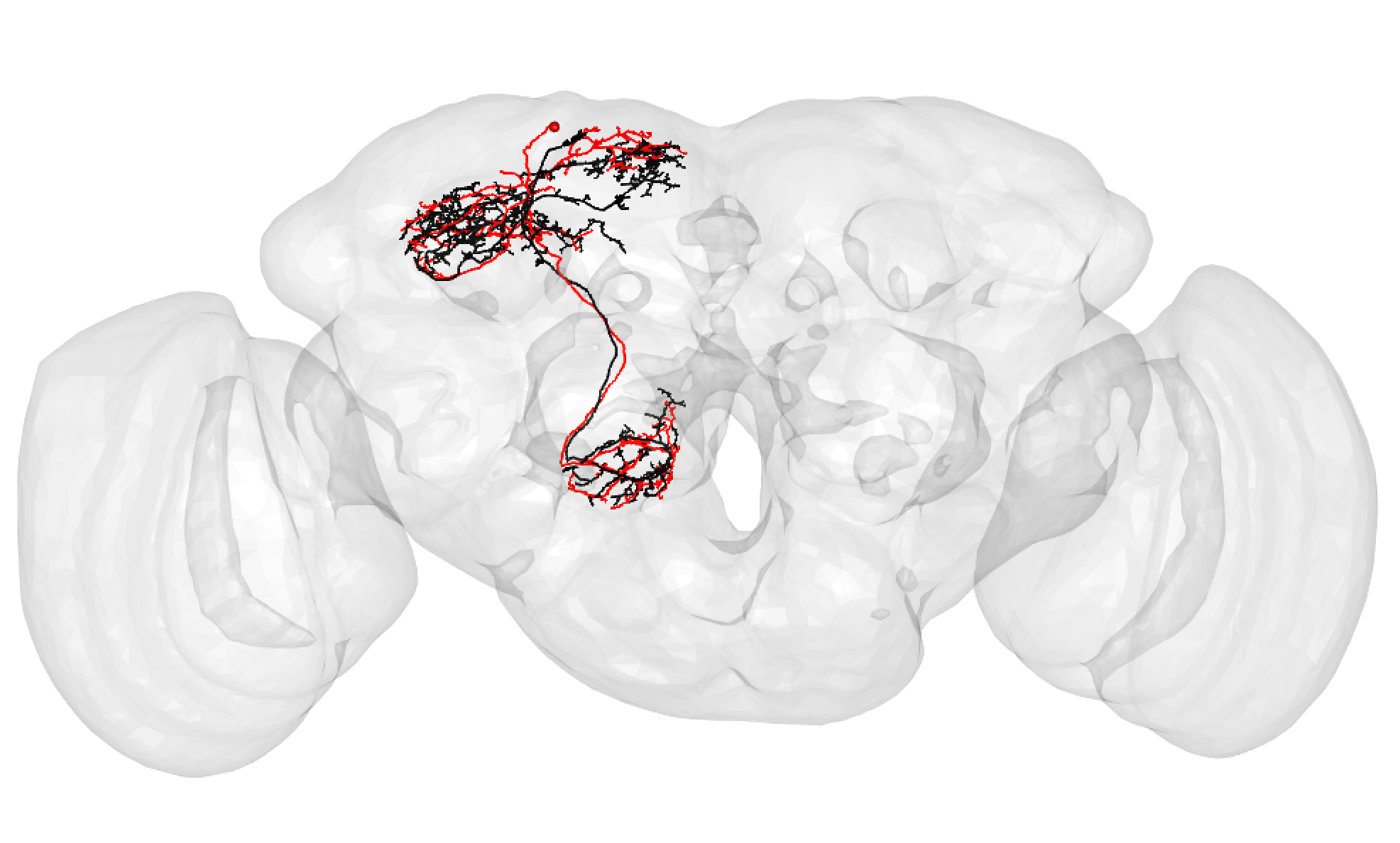
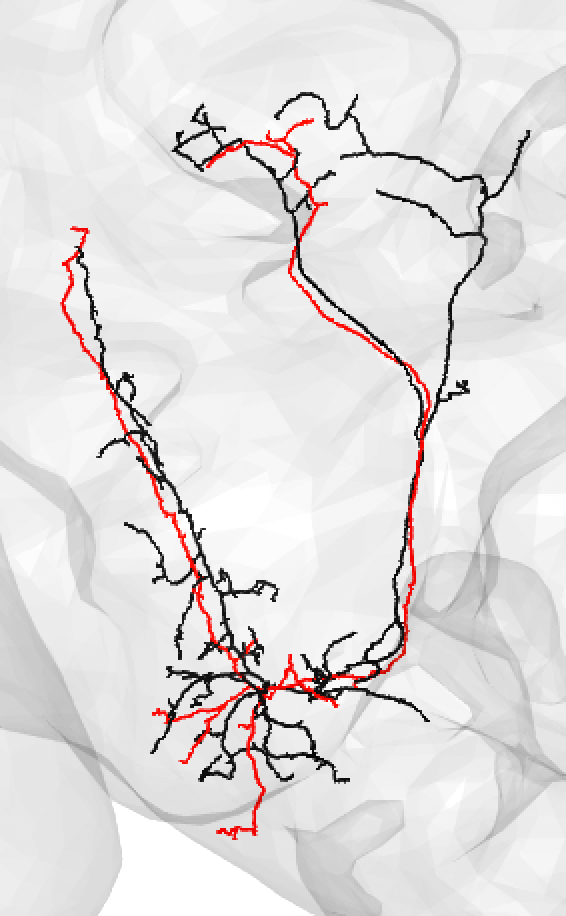
| Some good matches are striking. For example: | ||
|
|
||
|  | ||
|
|
||
| In the above case, the FAFB neuron has been quite extensively manually traced, meaning that these cells look very similar to one another. | ||
|
|
||
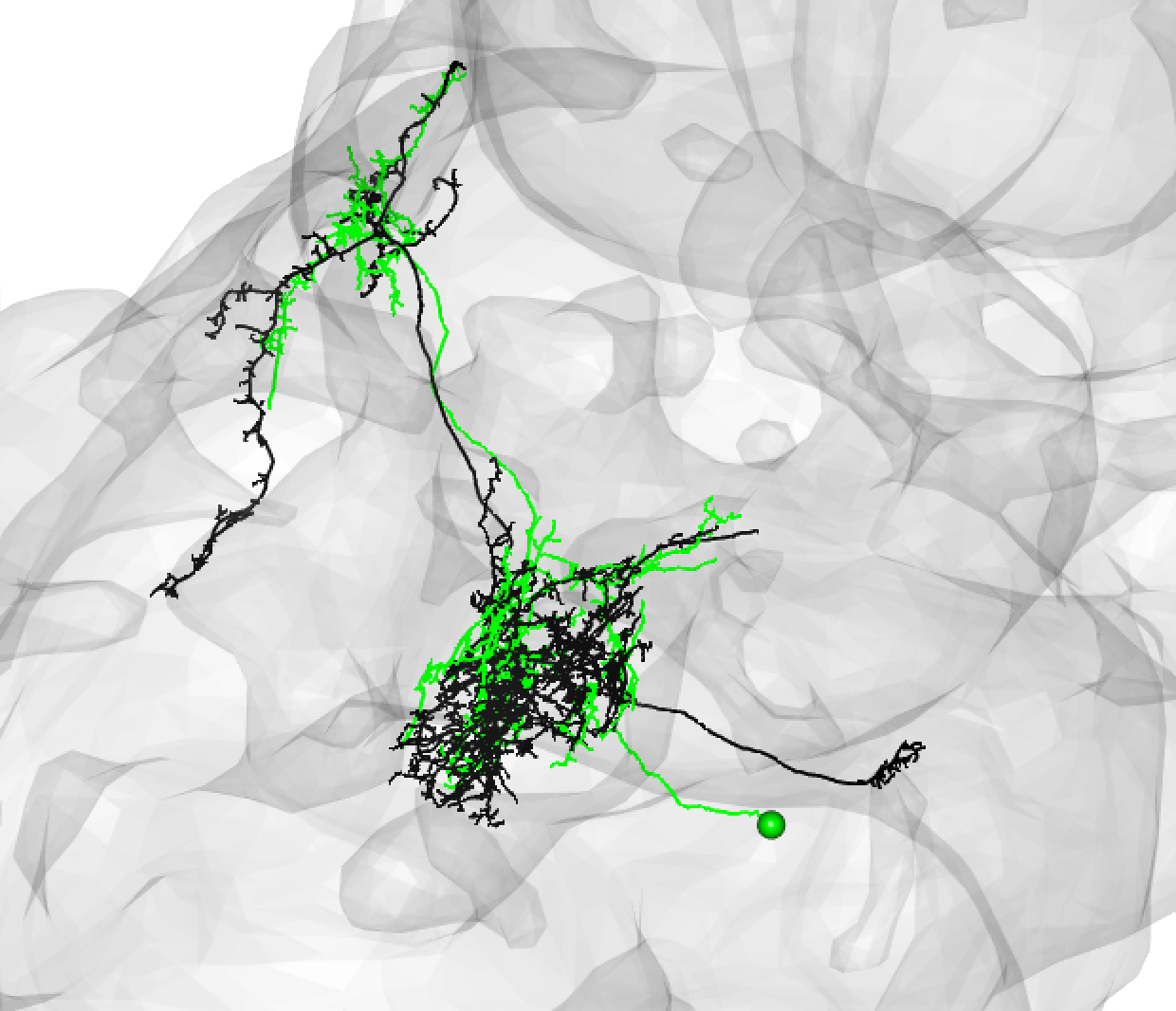
| Be aware that while neurons must share the same cel lbody fiber tract, these tracts can be a little off set. For example, this is also a good match: | ||
|
|
||
|  | ||
|
|
||
| If the some is missing, it might be safer to note a match as 'medium'. | ||
|
|
||
| 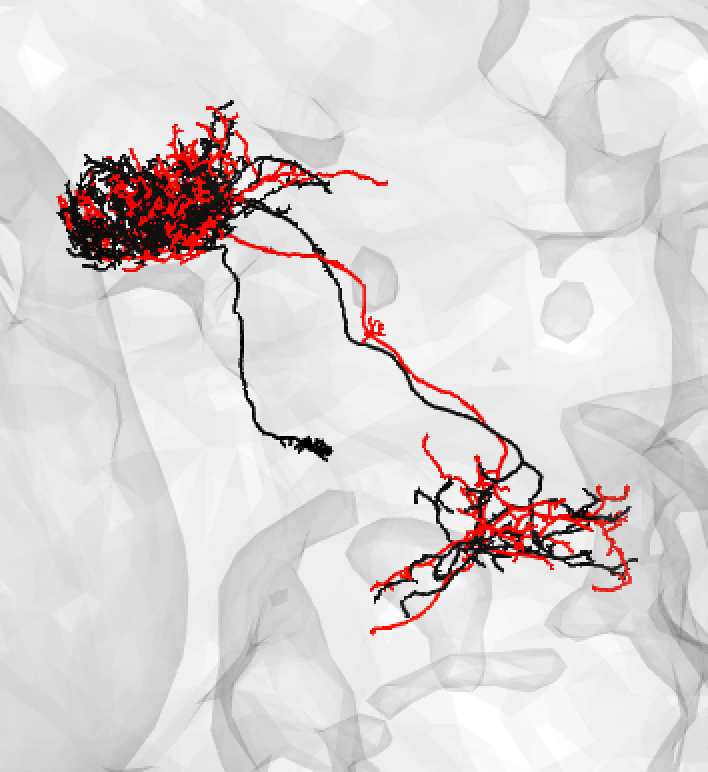 | ||
| You might also use medium if you have a nice looking match and suspect that there is a medium/large discrepancy because the FAFB neuron (here shown in red) is under-traced, such as: | ||
|
|
||
|  | ||
|
|
||
| Or: | ||
|
|
||
| 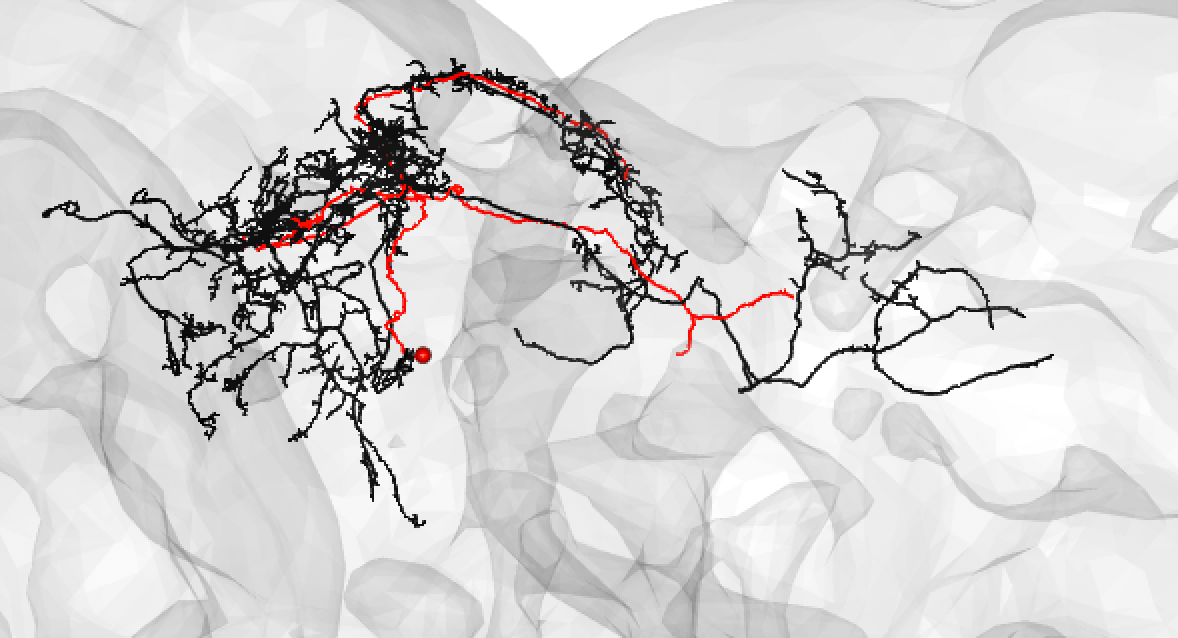 | ||
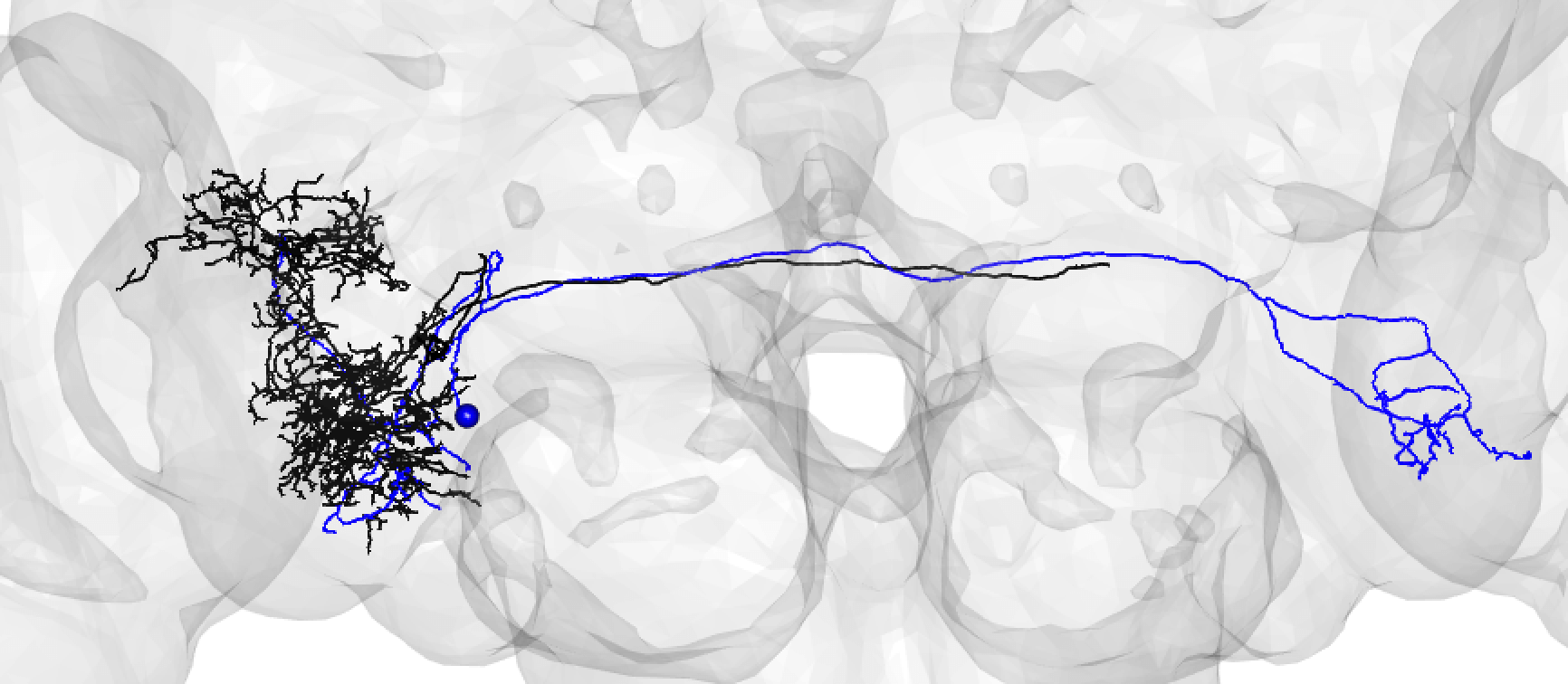
| Bear in mind that the hemibrain volume only covers ~1/4 of the fly mid-brain, so neurons are trunacted (here hemibrain neuron in black) but we can still make matches for many of them: | ||
|
|
||
|  | ||
|
|
||
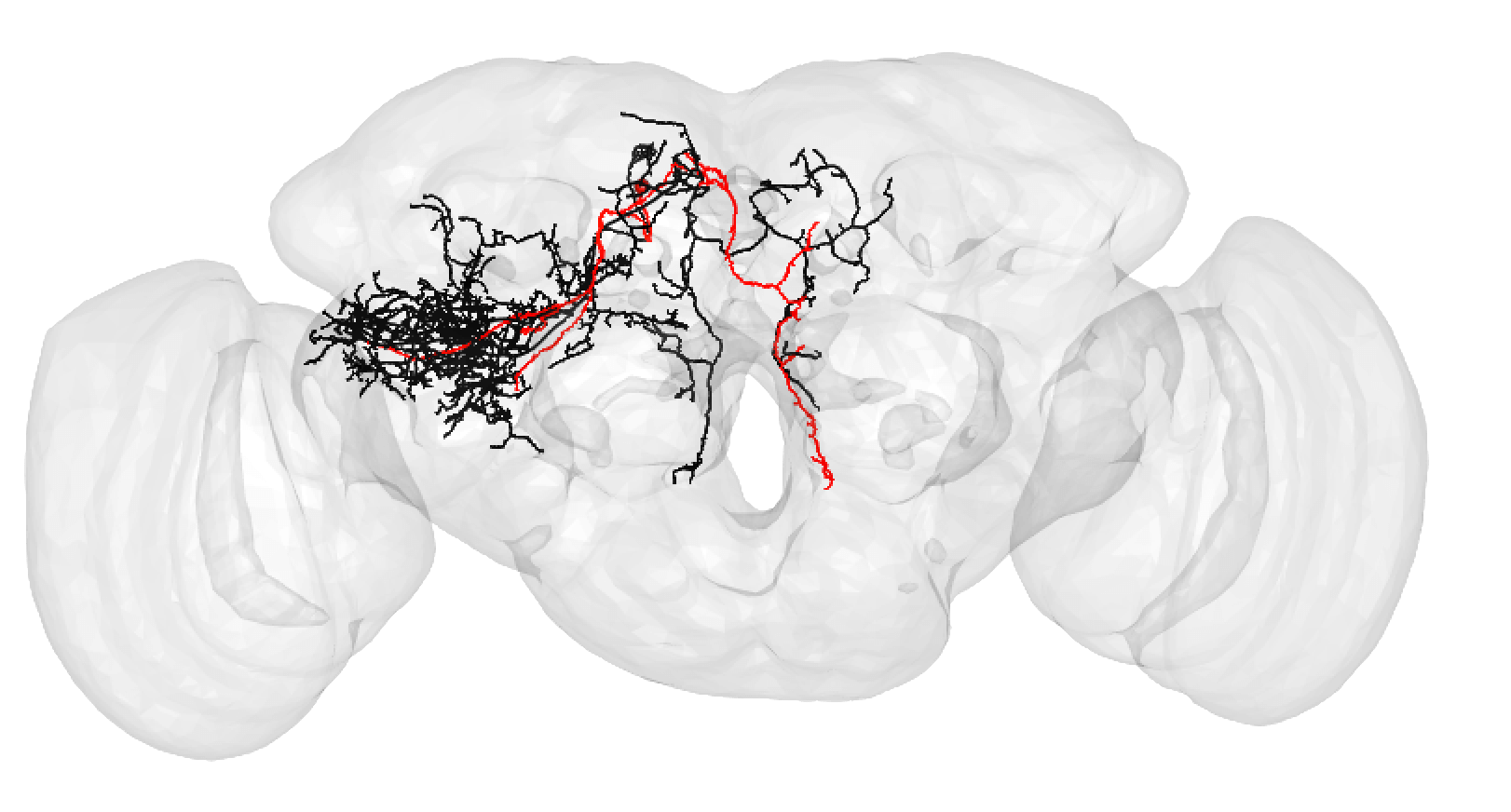
| A larger degree of under-tracing may lead you to assign a match as poor. In this case, you think the two neurons may be 'the same isomorphic cell type' but you could be wrong. For example: | ||
|
|
||
|  | ||
|
|
||
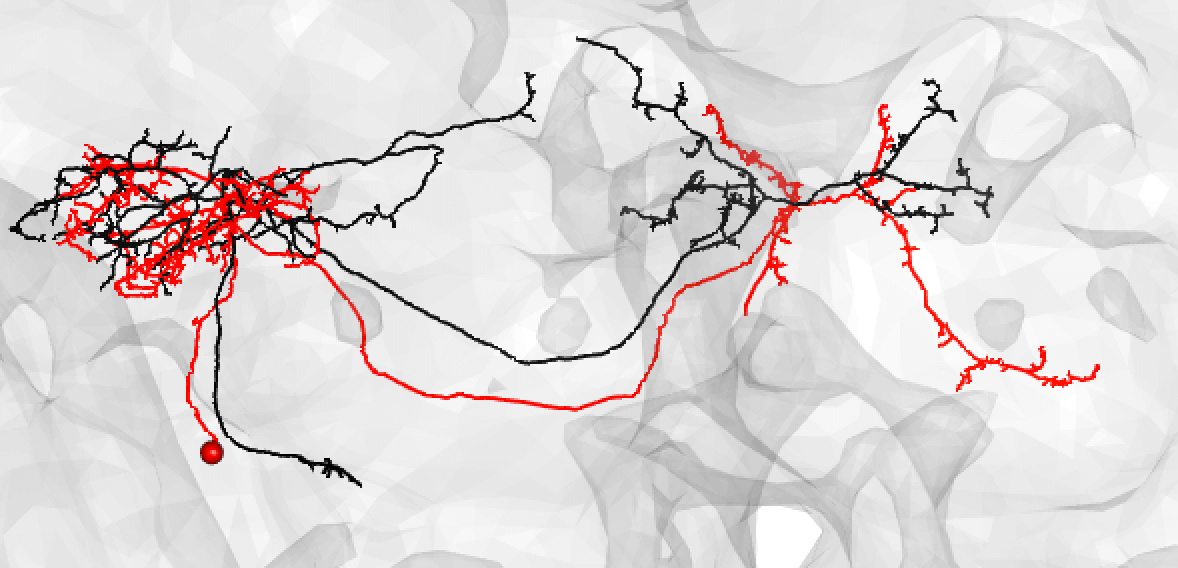
| A poor match may also be made if you think these is a slight offset, possibly due to a registration issue: | ||
|
|
||
|  | ||
|
|
||
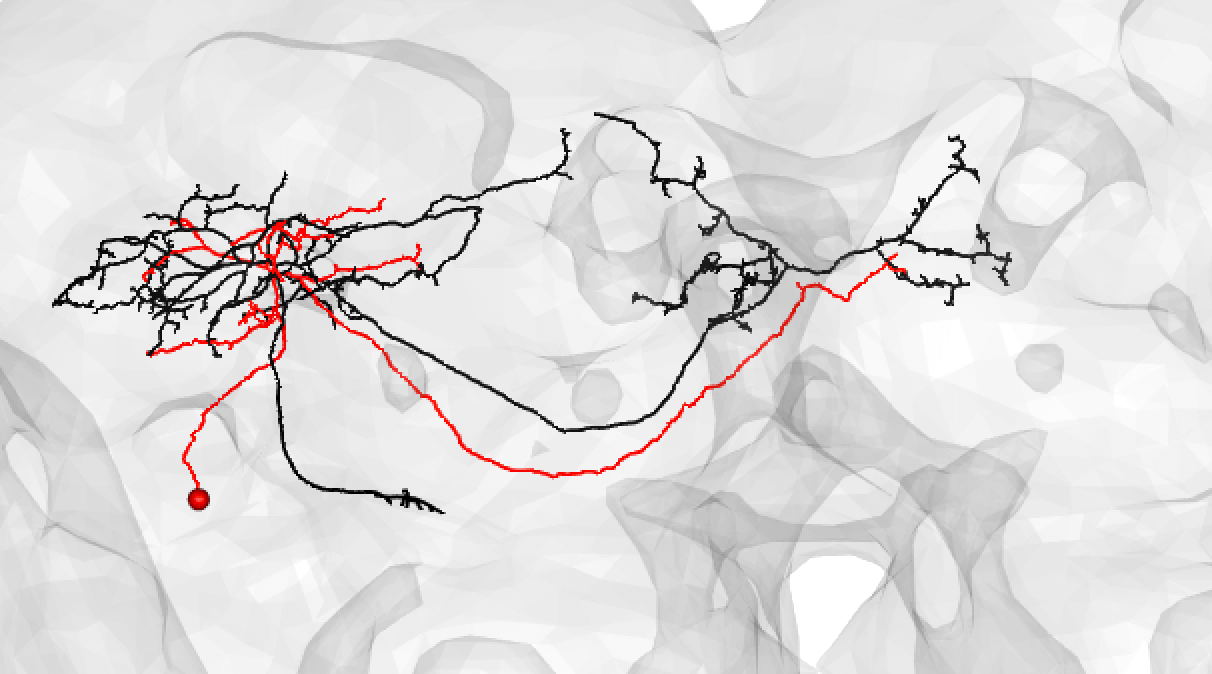
| Though in this case, choosing a even lesser-traced FAFB neuron may be better: | ||
|
|
||
|  | ||
|
|
||
| A poor match can be given even to very under-traced FAFB neurons: | ||
|
|
||
| 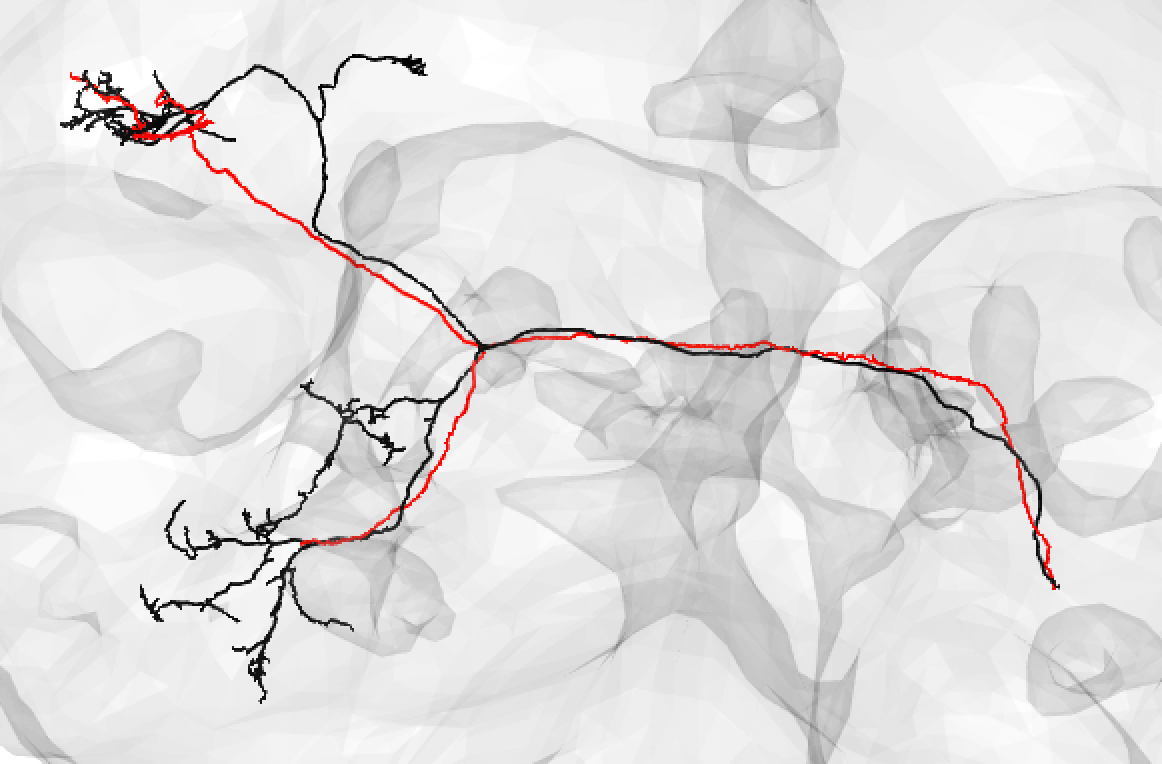 | ||
|
|
||
| And even fragments if you are convinced the morphology is unique enough (but be careful!): | ||
|
|
||
| 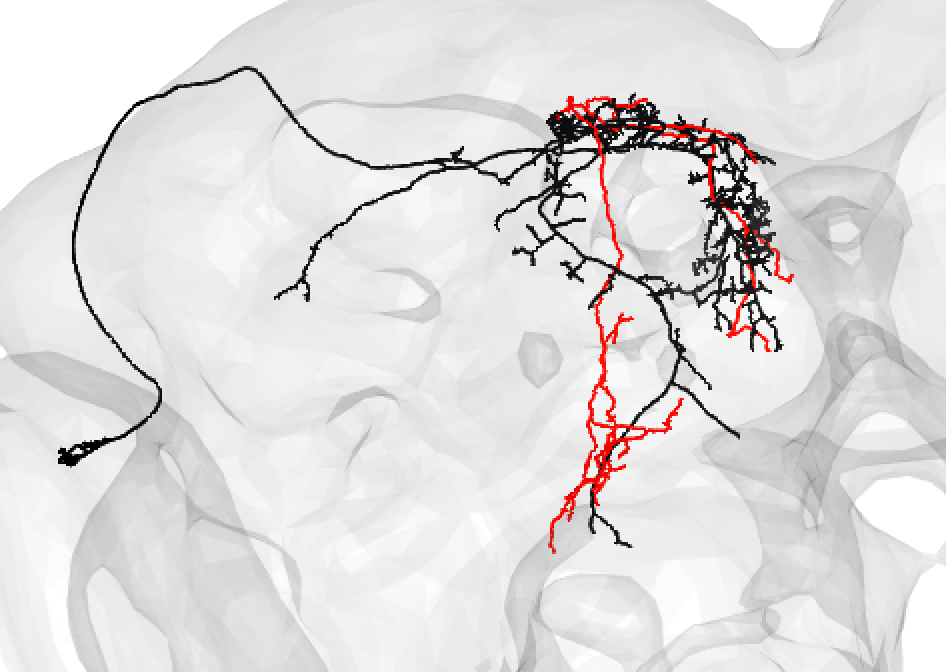 | ||
|
|
||
| ## Adding Your Own Matches | ||
|
|
||
| So far we have matched up a few thousand neurons. About 25 thousand matches are possible because that is the number of reconstructed neurons in the hemibrain data set. You can help us (and yourself!) by adding matches to our database. There are two main ways of doing this: | ||
|
|
||
| ### Adding Ready-Made Matches | ||
|
|
||
| You can add matches you have already made by your own means. For this, you will need to get a data frame into R (e.g. reading from a `.csv` file) that has three columns: `bodyid`, which contains the hemibrain neurons' unique Body IDs, `skid` which has the skeleton IDs for FAFB CATMAID neurons, and `quality` which gives a qualitative assessment of match quality (see above). If in doubt, put poor. | ||
|
|
||
| ```{r ready.made, eval = FALSE} | ||
| made.matches = read.csv("my_matches.csv") # Must have the named columns: bodyid, skid, quality | ||
| hemibrain_matches(df = made.matches, direction = "both") # direction controls which tabs matches get written to | ||
| ``` | ||
|
|
||
| Sometimes you cannot add a match, as your neuron either does not exist in the first column of the `hemibrain` tab of our Google sheet or of the `fafb` sheet. In these cases, if you have a valid ID, you can either add it to the sheet manually, or programmatically so that all the right meta data is easily included: | ||
|
|
||
| ```{r add, eval = FALSE} | ||
| # Add a mising FAFB projection neuron, so we can match it later: | ||
| hemibrain_matching_add(ids = "16", sheet = "fafb", User = "ASB") | ||
| ## the sheet argument specifies the worksheet or 'tab' on the Google sheet we want to add to | ||
| ``` | ||
|
|
||
| ### The Match Making Pipeline | ||
|
|
||
| You can also use our interactive pipeline to match neurons between hemibrain and FAFB. There are two version of this pipeline. One that takes hemibrain neurons from neuPrint, and tries to find the best match for each hemibrain neuron (`hemibrain_matching`) and one that takes FAFB neurons from CATMAID and tries to find the best heimibrain match for those FAFB neurons (`fafb_matching`). Once matches are made, the result become available with `hemibrain_matches`. | ||
|
|
||
| For a video tutorial, see [here](https://drive.google.com/drive/folders/1MOwfPGCPvY8eVLZoOfXOBhVcS6U996g9). | ||
|
|
||
| ```{r interactive.pipeline, eval = FALSE} | ||
| # install package to bridge neurons between FAFB14 and hemibrain space | ||
| if (!requireNamespace("remotes")) install.packages("remotes") | ||
| remotes::install_github('natverse/nat.jrcbrains') | ||
| nat.jrcbrains::download_saalfeldlab_registrations() | ||
| # Match hemibrain neurons! | ||
| hemibrain_matching() # Automatically, you can choose a User ID and you are given neurons that have this ID in the User column on the Google Sheet. | ||
| hemibrain_matching(ids=c("674108632","739256609")) # Otherwise you can select specific IDs | ||
| hemibrain_matching(ids=c("674108632","739256609"), overwrite = TRUE) # If a match has already been made you can overwrite it | ||
| # Otherwise neurons that have already been given a match will not be shown in the pipeline. | ||
| # Match FAFB neurons! | ||
| fafb_matching() | ||
| fafb_matching(ids = "16") # Specify IDs | ||
| fafb_matching(ids = "16", overwrite = TRUE) # Overwrite | ||
| ``` | ||
|
|
||
| When you run these functions you will enter an interactive pipeline in an rgl window. Prompts will be given to you in your R console and you can rotate and pan in the window to see neurons. The neuron selected for-matching is shown in black (i.e. if using `hemibrain_matching` this will be a hemibrain neuron), and potential matches in colour (i.e. if using `hemibrain_matching` these will be FAFB neurons). Potential matches are shown by NBLAST score (a measure of morphological similarity). Usually, for reasonably traced FAFB neurons, a good match appears in the top 10 hits. | ||
|
|
||
|  | ||
|
|
||
| ## Uses | ||
|
|
||
| One use we have already found for all of this match making, is to cross-identify neuron cell body fiber tracts and [(hemi)lineages](https://pubmed.ncbi.nlm.nih.gov/23541733/). This means that we now have the locations in FAFB for different known sets of cells. You can see seed planes for them [here](https://docs.google.com/spreadsheets/d/1HI8RZJhV8aWC6hw5T0qh2__9D8V0zKdtxx2kkDsuI8Y/edit?usp=sharing). | ||
|
|
||
| 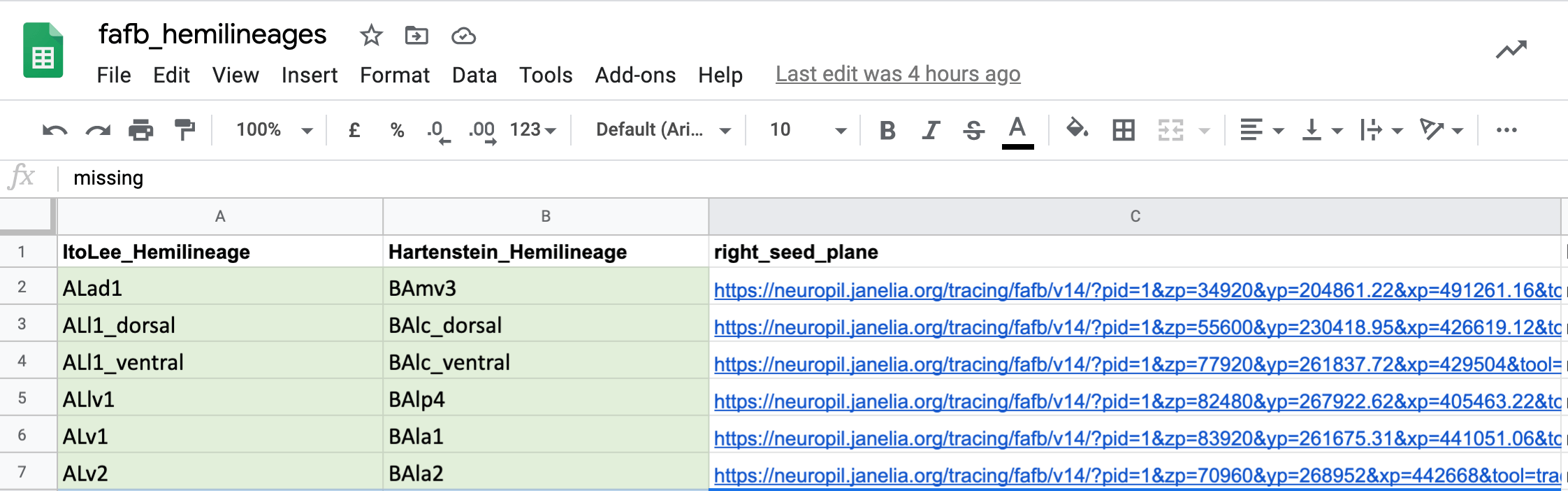 | ||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
Oops, something went wrong.