- + hemibrain and flywire meta data + +
- flywire
-
@@ -108,9 +111,6 @@
flywire
--
“We’re Building Something, Here, Detective, We’re Building It From Scratch. All The Pieces Matter.” (Lester, The Wire)
-FlyWire and R
@@ -122,7 +122,7 @@- Read flywire NBLASTs and NBLASTs to hemibrain neurons
- Read flywire neurons that are pre-transformed into a variety of brainspaces
-Which is all useful stuff. In order to connect R to this Google drive, you have a few options. Please see this article. The pipeline that produced this data can be found here. It is run nightly on a machine at the MRC LMB.
+Which is all useful stuff. In order to connect R to this Google drive, you have a few options. Please see this article. The pipeline that produced this data can be found here. It is run nightly on a machine at the MRC LMB.
-+diff --git a/docs/articles/google_filestream.html b/docs/articles/google_filestream.html index 55e916dd..bfb0edef 100644 --- a/docs/articles/google_filestream.html +++ b/docs/articles/google_filestream.html @@ -55,6 +55,9 @@-Get flywire
+Get flywire dataWith
hemibrainryou can easily get thousands of skeletons for flywire neurons. These skeletons have been built using skeletor, which is a python module. You can also use it directly in R withfafbseg::skeletor. It has been run on thousands of neurons, which have then been stored on Google Drive, at:hemibrainr/flywire_neurons/asnat::neuronlistfhobjects. This means that you can use either get flywire skeletons for neurons using the drive, or by making them from meshes yourself. Let’s demonstrate:@@ -172,7 +172,7 @@
From the the hemibrainr Google drive we can see which flywire IDs have been read from FlyWire and skeletonised. We can also see some meta data about them, and get a data frame that tells us which users have built these neurons - useful for assigning credit!
+head(fw.edits) + +# For flywire IDs which do not have available skeletons on the google drive: +## but have been flaghged for processing (i.e. they errored) +fw.failed = flywire_failed() +head(fw.failed)# All flywire IDs for neurons that have a split precomputed -fw.ids = flywire_ids() +fw.ids = flywire_ids(sql=FALSE) length(fw.ids) # For these flywire IDs, their meta data: @@ -181,7 +181,12 @@# For flywire IDs, which users contributed what: fw.edits = flywire_contributions() -head(fw.edits)
Now, excitingly we can read the neurons themselves! And not only that, we can read them mirrored (flipped to the other hemisphere of the brain) or in a different brainspace (from a small selection) if we wish. We read these neurons as
nat::neuronlistfhobjects. This means that a data frame for the neurons and information specifying where to find each neuron’s data is read into R - but not the whole, hugenat::neuronlistobject. This saves on memory for your R session. When an operation that requires the actual neuron data is performed, neurons are read into R.+# Get all aflywire neurons @@ -281,7 +286,7 @@Match flywire neurons to each other and the hemibrain
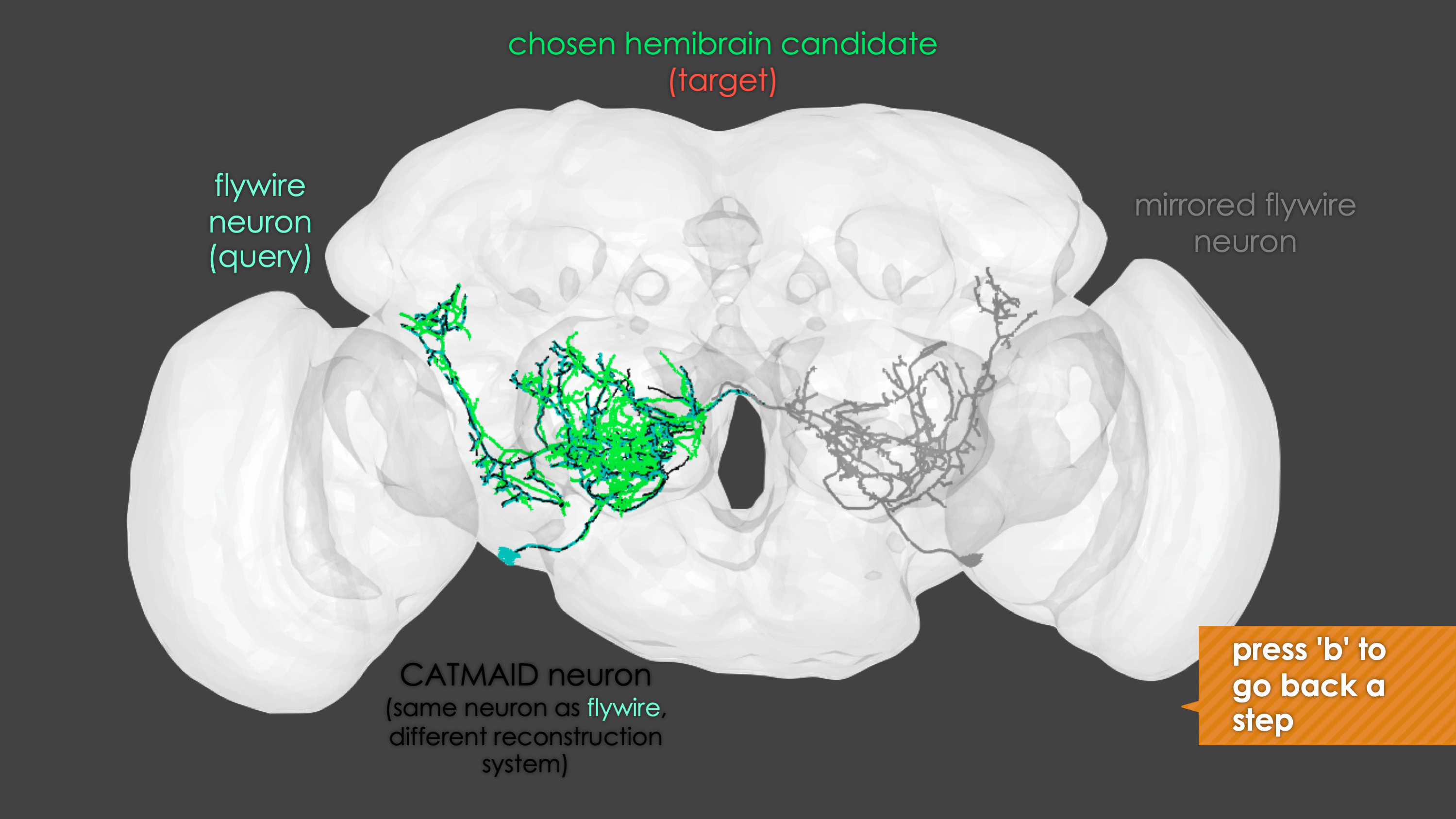
-The neuron matching pipeline reads neuron skeletons from Google Drive and and displays them. FlyWire skeletons are shown in dark grey (their mirrored equivalents in light grey) and potential hemibrain matches in red. This depends also on the precomputed NBLASTs on the hemibrain team drive. For full details, see our
+match_makingvignette.The neuron matching pipeline reads neuron skeletons from Google Drive and and displays them. FlyWire skeletons are shown in dark grey (their mirrored equivalents in light grey) and potential hemibrain matches in red. This depends also on the precomputed NBLASTs on the hemibrain team drive. For full details, see our
match_makingvignette.Matching pipelines
@@ -309,6 +314,26 @@flywire_matching_rewrite() # bring all the flywire information up to date++Full Example
+Let us get some neurons from the ‘flywire interest’ googlesheet:
+++
# Get all the flywire IDs (updated regularly based on position information) users have flagged +gs = googlesheets4::read_sheet(ss = options()$flywire_flagged_gsheet, sheet = "flywire") + +# Get IDs for User 'Tots' +gs.chosen = subset(gs, User == "Tots") + +# Load processed flywire neurons +fw = flywire_neurons() + +# Get those for Tots +fw.tots = fw[names(fw)%in%gs.chosen$flywire.id] + +# Examine their meta data +fw.tots[,]- + hemibrain and flywire meta data + +
- flywire
-
@@ -124,6 +127,9 @@
The best option is to use google filestream. By default, this is what
hemibrainrexpects. However, you need a Google Workspace account (formerly G-Suite), which is a paid-for service. Without this, the best option is to use rclone. You can also download to an external hard drive and use that.We have two Google team drives available for you to use, which contain similar data. One (
+"hemibrain") is for internal use by the Drosophila Connectomics Group. The other one ("hemibrainr") is shared with those who would like access. Contact us by email to request access.+ 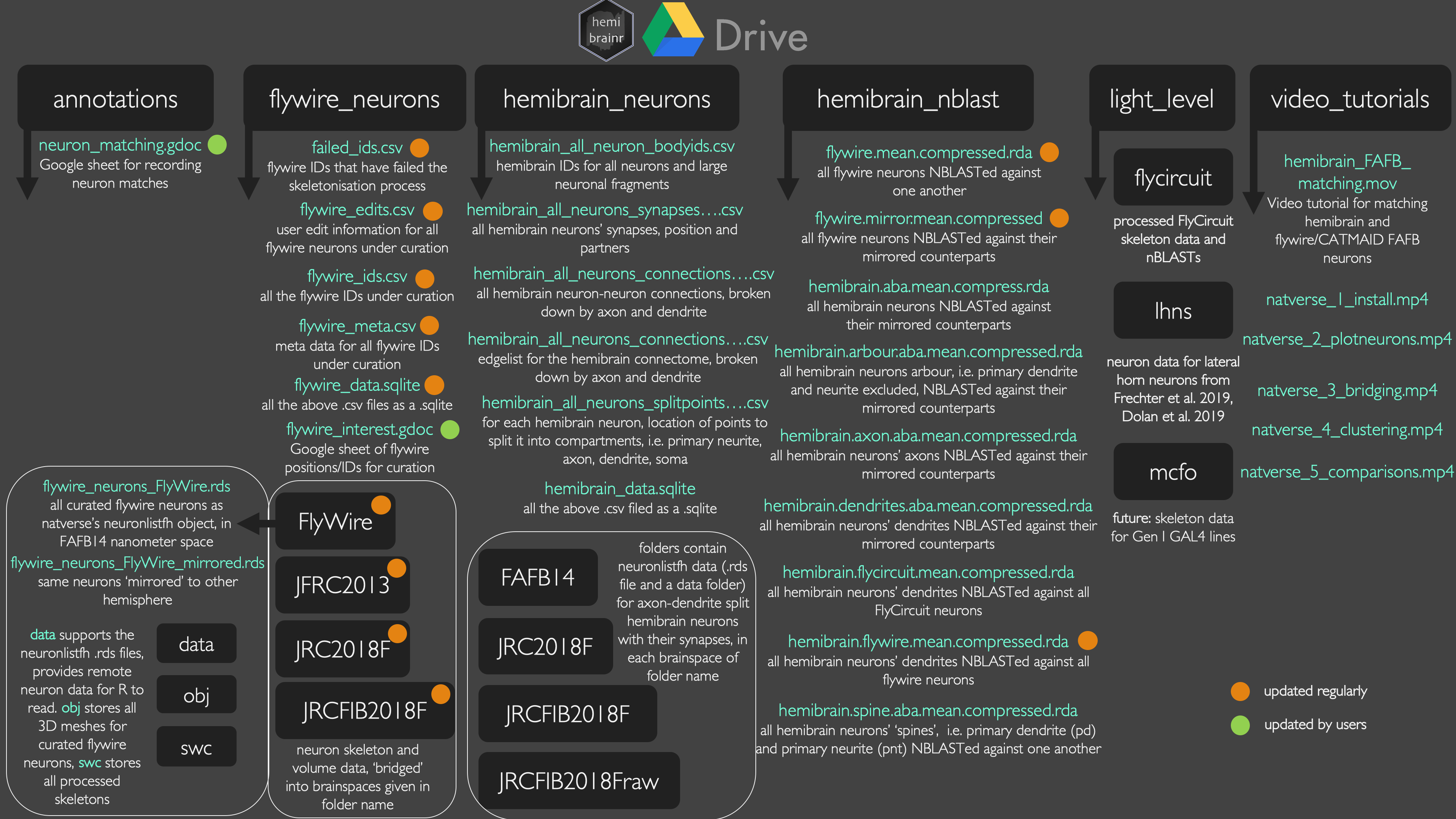 +@@ -194,7 +197,7 @@
+@@ -194,7 +197,7 @@Option 1: Google workspace, hemibrainr and R
@@ -162,7 +168,7 @@Set your drive location
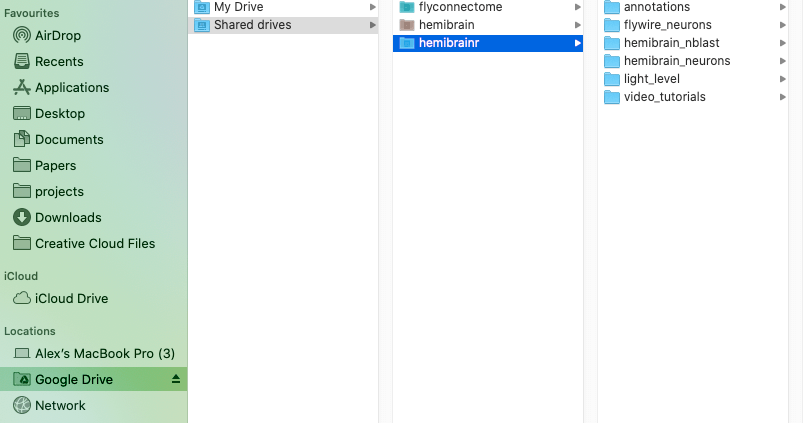
If you have mounted your Google drives with Google file stream, you should be able to see something like this:
- 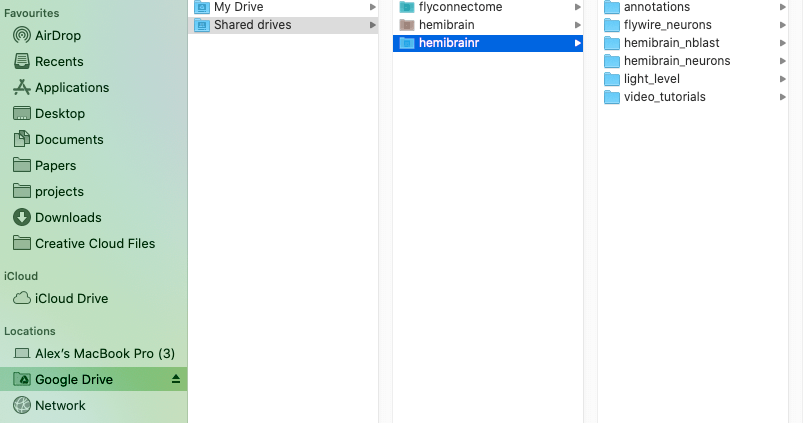 +
+
If you need to set
hemibrainrto look at a new drive, use:@@ -270,7 +276,7 @@
Once you have installed and configured rclone, you can use it to mount the google team drive. To do this, you create an empty directory wherever you want, named whatever you like, and then rclone ‘turns’ this directory into your mounted Google drive.
You can mount with rclone from your system’s command line:
+rclone mount hemibrainr: /path/to/local/mountTry it somewhere like
/Documentsto see how it works. Forhemibrainrthere is an R function that will do the mounting for you in your working directory. So within R:diff --git a/docs/articles/hemibrain_axons_dendrites.html b/docs/articles/hemibrain_axons_dendrites.html index a44a4b82..2e2d6d0c 100644 --- a/docs/articles/hemibrain_axons_dendrites.html +++ b/docs/articles/hemibrain_axons_dendrites.html @@ -55,6 +55,9 @@# mounts in working directory diff --git a/docs/articles/hemibrain_alpns_toons.html b/docs/articles/hemibrain_alpns_toons.html index ab067925..8b9a73c8 100644 --- a/docs/articles/hemibrain_alpns_toons.html +++ b/docs/articles/hemibrain_alpns_toons.html @@ -55,6 +55,9 @@- + hemibrain and flywire meta data + +
- flywire
-
@@ -161,12 +164,12 @@
Get neuron connectivity data
-Now we can get a precomputed edgelist for the brain from the Google drive, which breaks down connections by axon and dendrite. By default this is read from an SQLlite database on the Google drive. This means we can access the data, without loading a multi Gb .csv into memory:
+Now we can get a precomputed edgelist for the brain from the Google drive, which breaks down connections by axon and dendrite. By default this is read from an SQLite database on the Google drive. This means we can access the data, without loading a multi Gb .csv into memory:
-# Get the edgelist elist = hemibrain_elist() meta = rbind.fill(ton.info, pn.info)In our
+elist, count is the number of synaptic connections between two arbours. The arbour types are given byLabelandpartner.Label. The neuron identities are given aspre, the upstream (source) neuron andpost, the downstream (target) neuron. The value ofnormis generated by takingcountand dividing it by the total number of posynapses on the downstream target’s given arbour (note, not all connection in the neuron!). See your article on neuron splitting, for more information.In our
elist, count is the number of synaptic connections between two arbours. The arbour types are given byLabelandpartner.Label. The neuron identities are given aspre, the upstream (source) neuron andpost, the downstream (target) neuron. The value ofnormis generated by takingcountand dividing it by the total number of postsynapses on the downstream target’s given arbour (note, not all connection in the neuron!). See your article on neuron splitting, for more information.alpn.lhn.elist <- elist %>% filter(pre %in% local(pn.info$bodyid) @@ -199,7 +202,7 @@} conn.mat.scaled = t(apply(conn.mat,1,normalise))
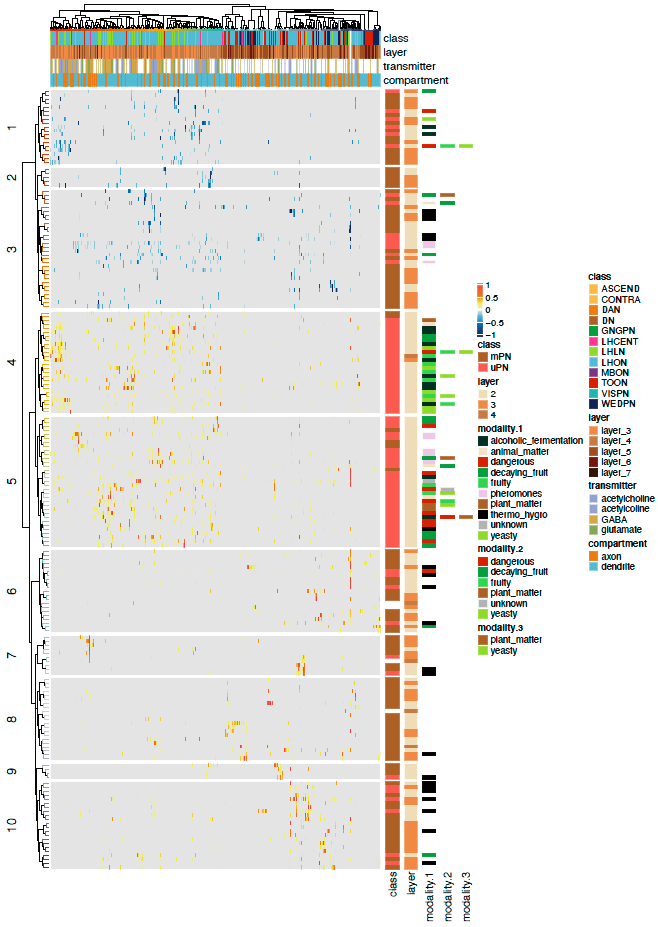
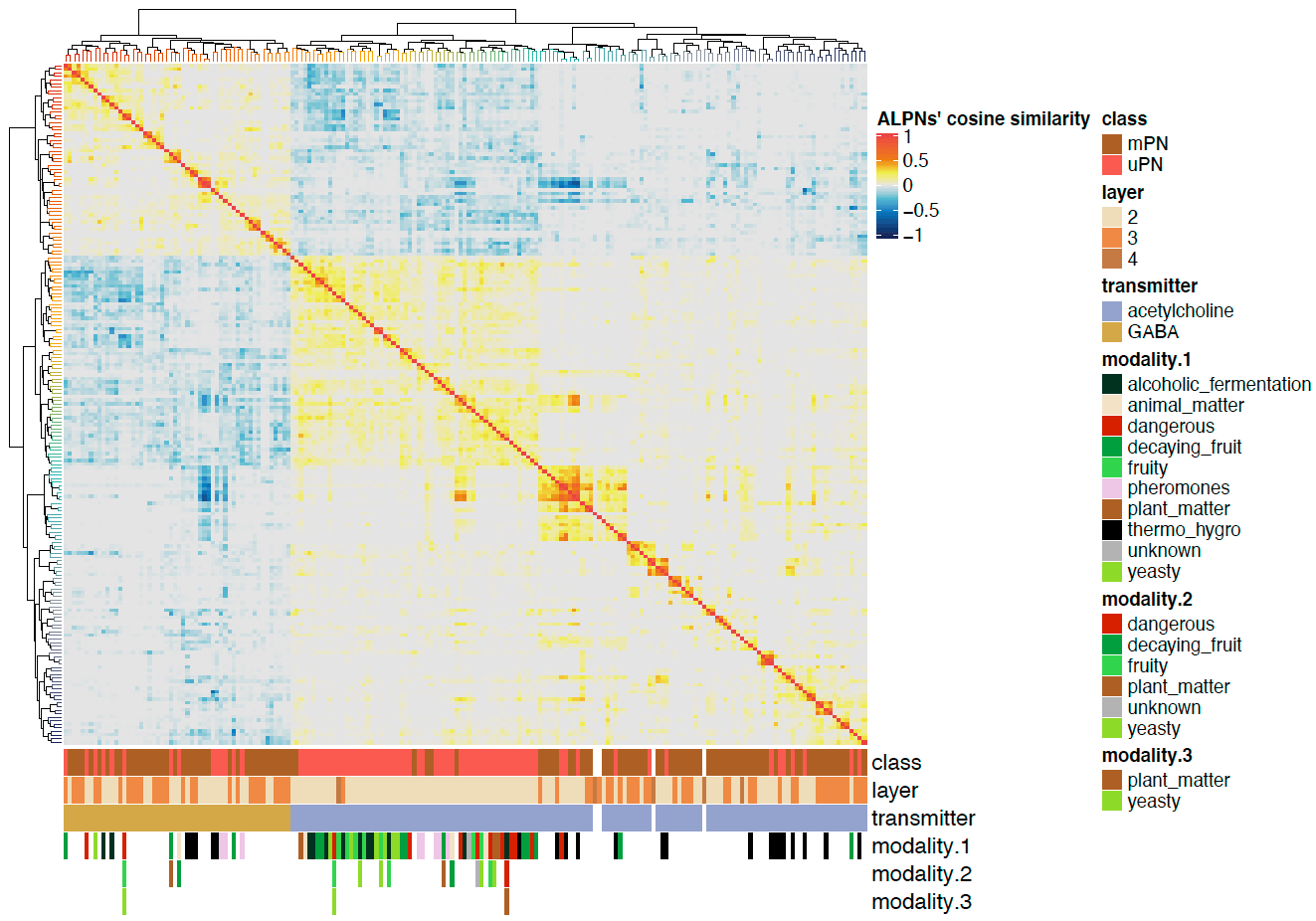
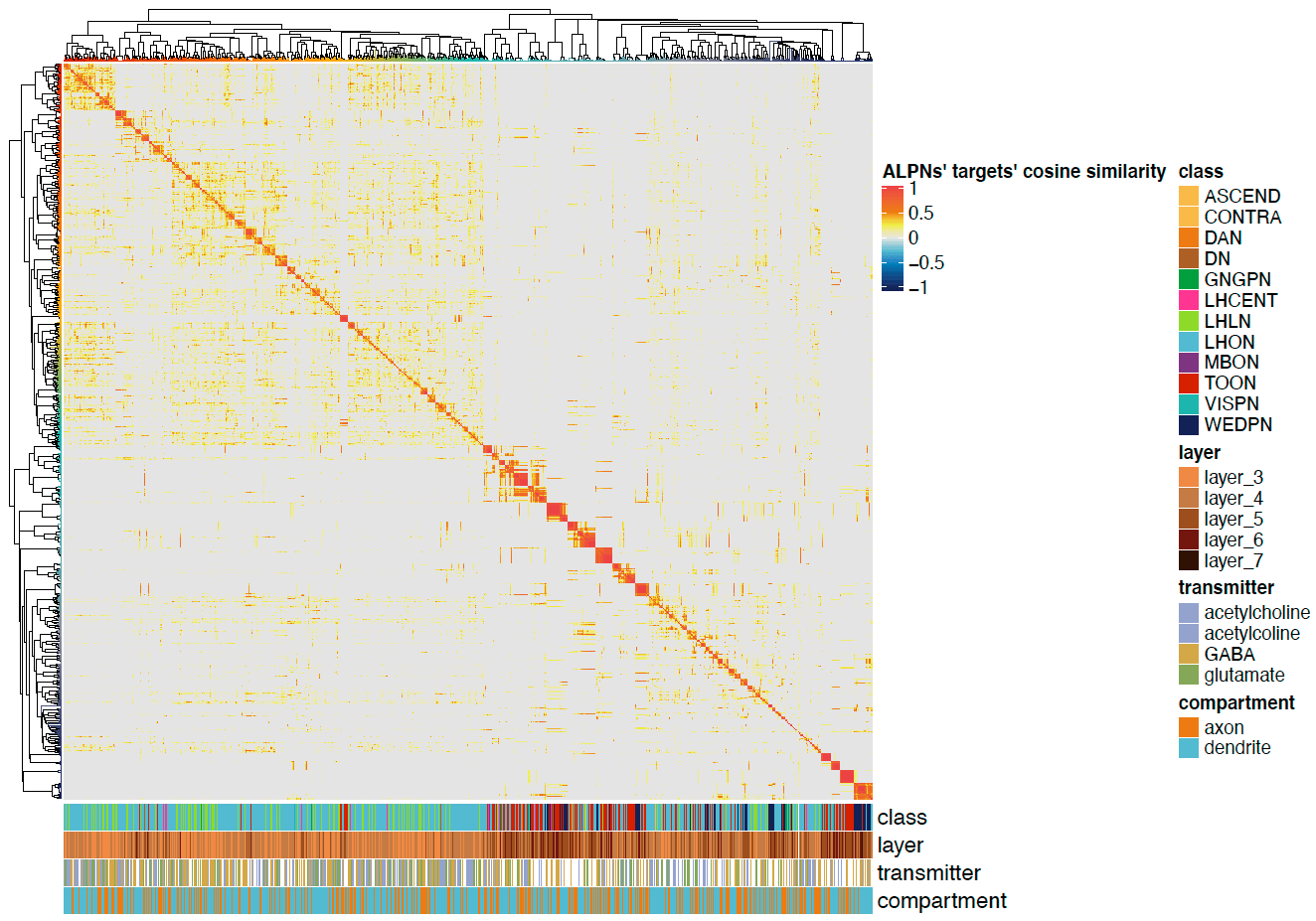
Now we can plot a heatmaps of the ALPN->TON weights, and correlation heatmaps.
-First let us get some clusterings for ALPNs and TONs based on ther cross-correlation.
+First let us get some clusterings for ALPNs and TONs based on their cross-correlation.
# TON correlation matrix cor.mat = lsa::cosine(conn.mat.scaled) @@ -340,7 +343,7 @@column_names_gp = gpar(fontsize = 12, fontfamily ="Helvetica")) dev.off()
- 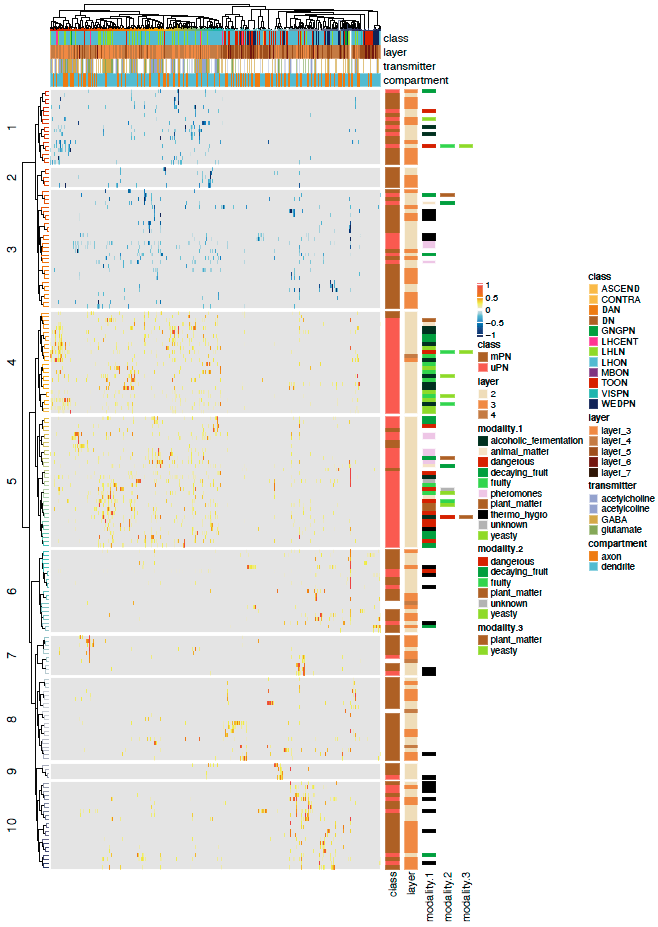 +
+
We can also plot the correlation heatmaps.
@@ -379,10 +382,10 @@
) dev.off()
- 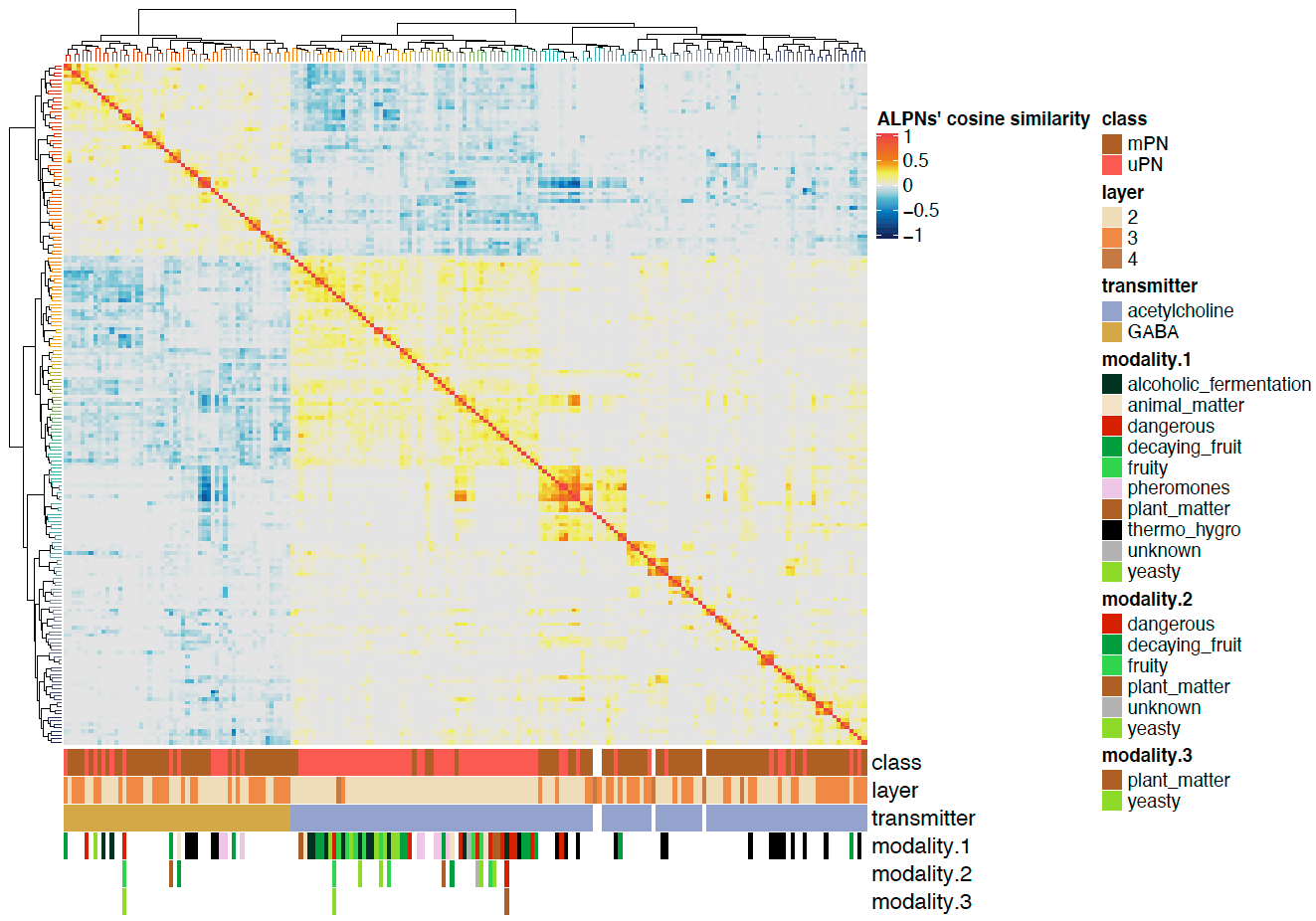 +
+
- 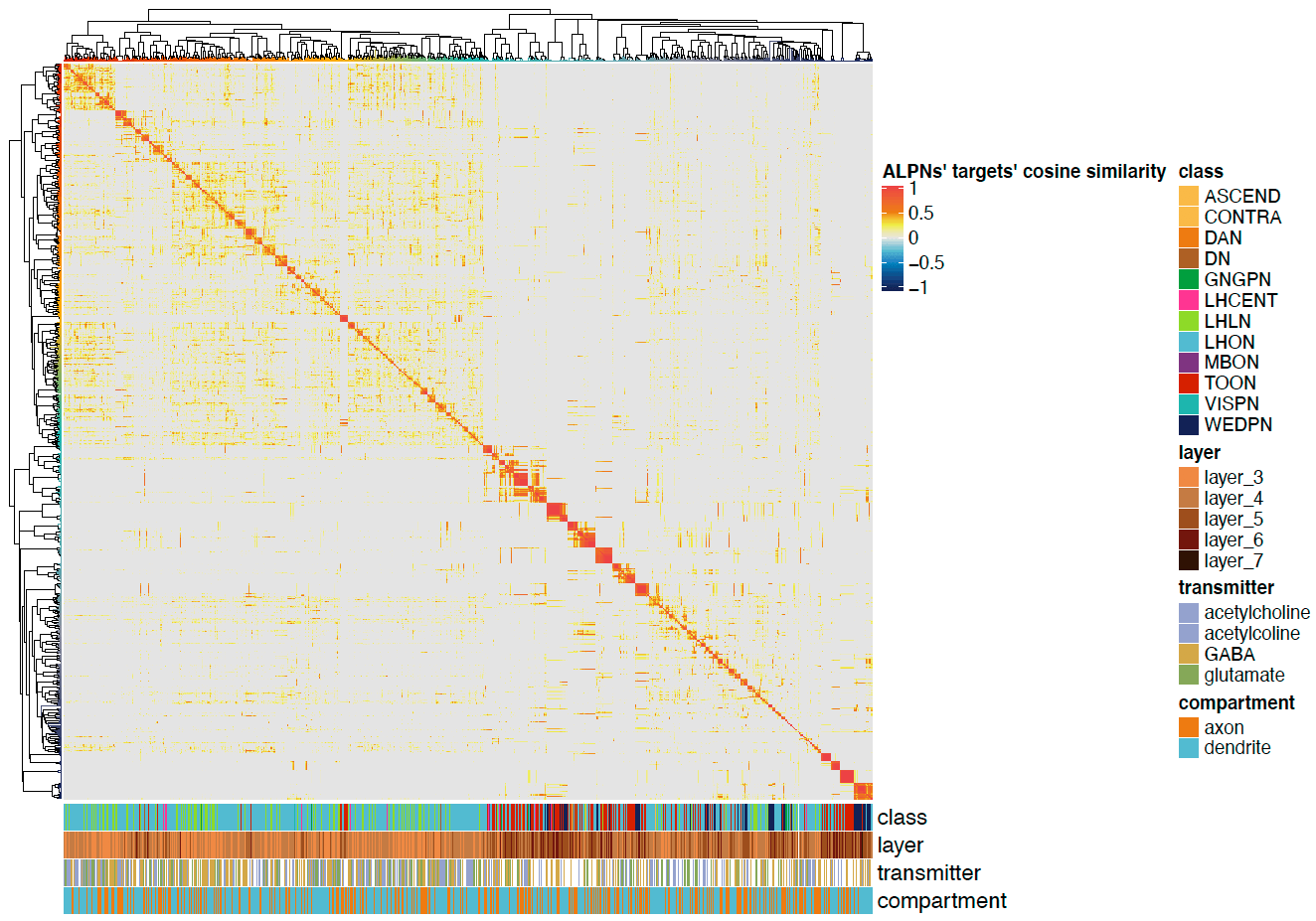 +
+
- + hemibrain and flywire meta data + +
- flywire
-
@@ -121,12 +124,12 @@
Splitting a neuron
Neurons can be split into a principle input zone, a dendrite, and a principle output zone, an axon. Dendrites tend to be more spindly and axons bulkier, with boutons packed with presynapses, ready to release neurotransmitter.
- 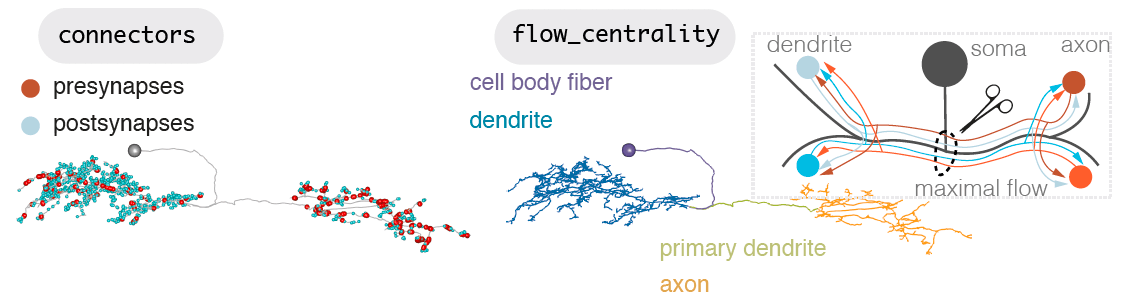 +
+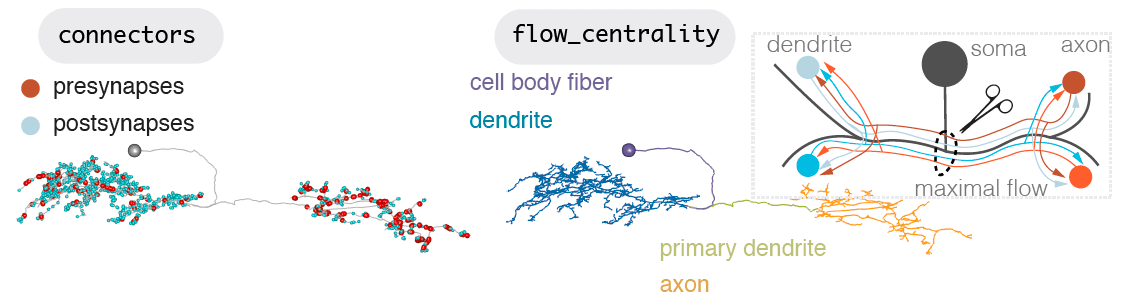
But axons and dendrites are not a totally clear division for most neurons. A neuron may have more than one of each. It may also be more ‘amacrine-like’ and lack this division, though this is less common. Dendrites can have outputs, and axons can have inputs. Axons may connect with axons, even dendrites may input and axon.
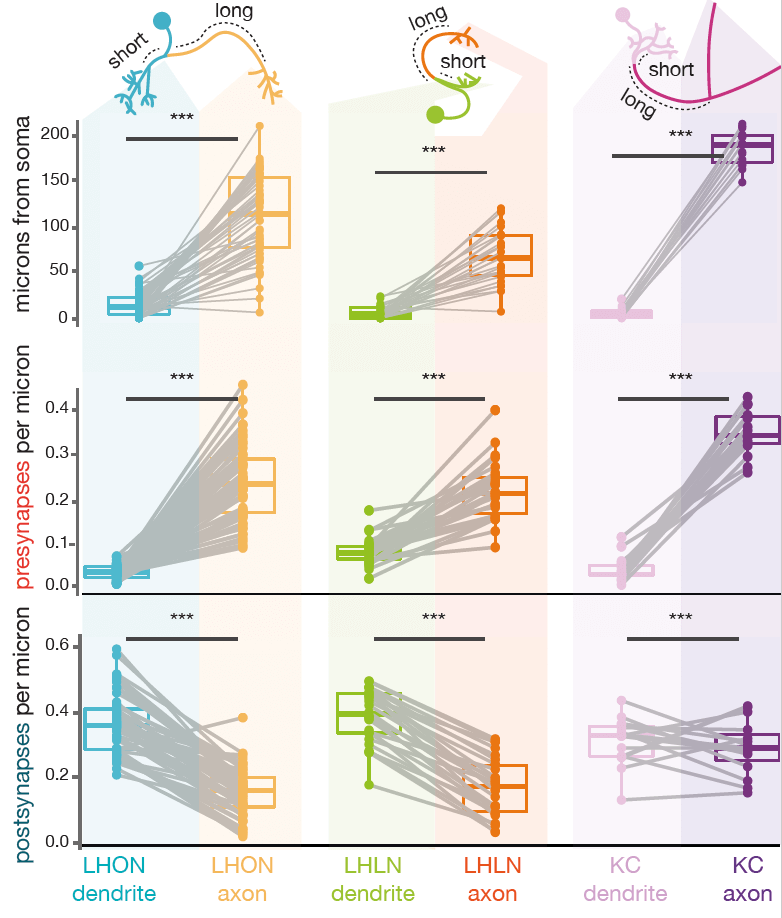
However, all fly neurons examined can be split into an axon-like and a dendrite-like compartment (Schneider-Mizell et al., 2016)). The density of outputs in the axon is higher than in the dendrite. The density of inputs to the dendrite, are higher than in the axon (Bates & Schlegel et al., 2020)). Interestingly, the ‘start’ of a dendrite is almost always closer to the neuron’s cell body than the start of its axon. See our recent data (Bates & Schlegel et al., 2020)) for three broad classes of olfactory neuron:
- 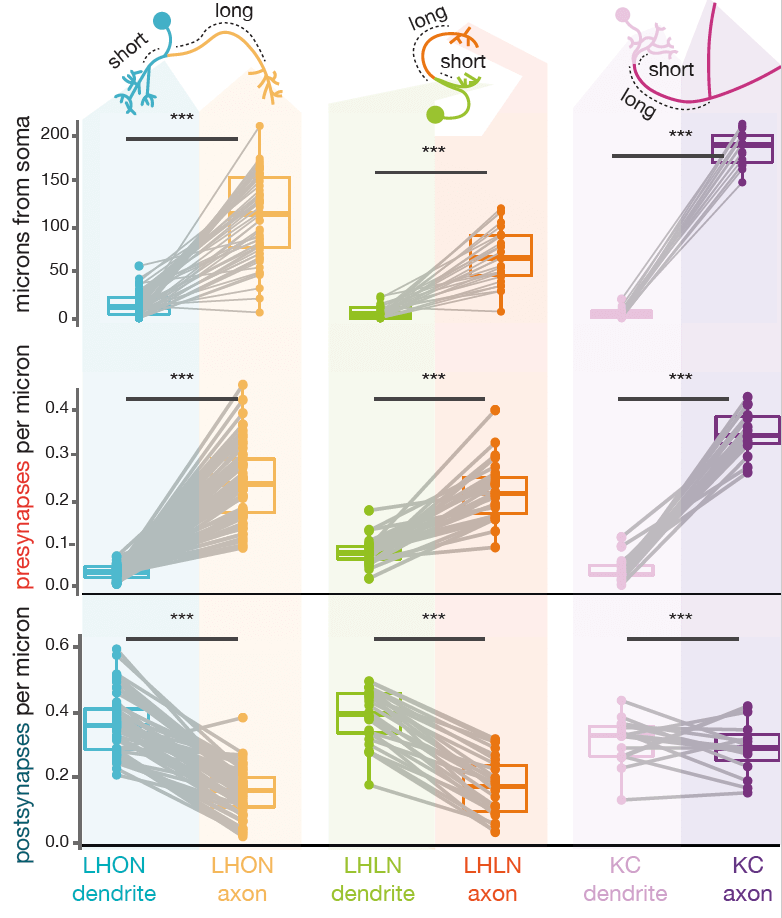 +
+
The biophysical impact of axo-axonic connections is unclear – they can have non-intuitive effects depending on the timing of action potentials in connected partners (Burrows and Laurent, 1993; Burrows and Matheson, 1994; Cuntz et al., 2007; Haag and Borst, 2004).
Get already split neurons
-Splitting neurons can take a little time. We can also load thousands of pre-split neurons from the
+hemibrainrGoogle team drive. In order to connect R to this Google drive, you have a few options. Please see this article. Once you have access and have mounted the drive, you should be able to do:Splitting neurons can take a little time. We can also load thousands of pre-split neurons from the
hemibrainrGoogle team drive. In order to connect R to this Google drive, you have a few options. Please see this article. Once you have access and have mounted the drive, you should be able to do:# All split hemibrain neurons db = hemibrain_neurons() @@ -234,7 +237,7 @@axon.synapses = hemibrain_extract_synapses(axons, prepost = "PRE") head(axon.synapses)
-  +
+ @@ -248,7 +251,7 @@
@@ -248,7 +251,7 @@plot3d_split(neurons.flow) plot3d(hemibrain.surf, col = "lightgrey", alpha = 0.1, add = TRUE)
-  +
+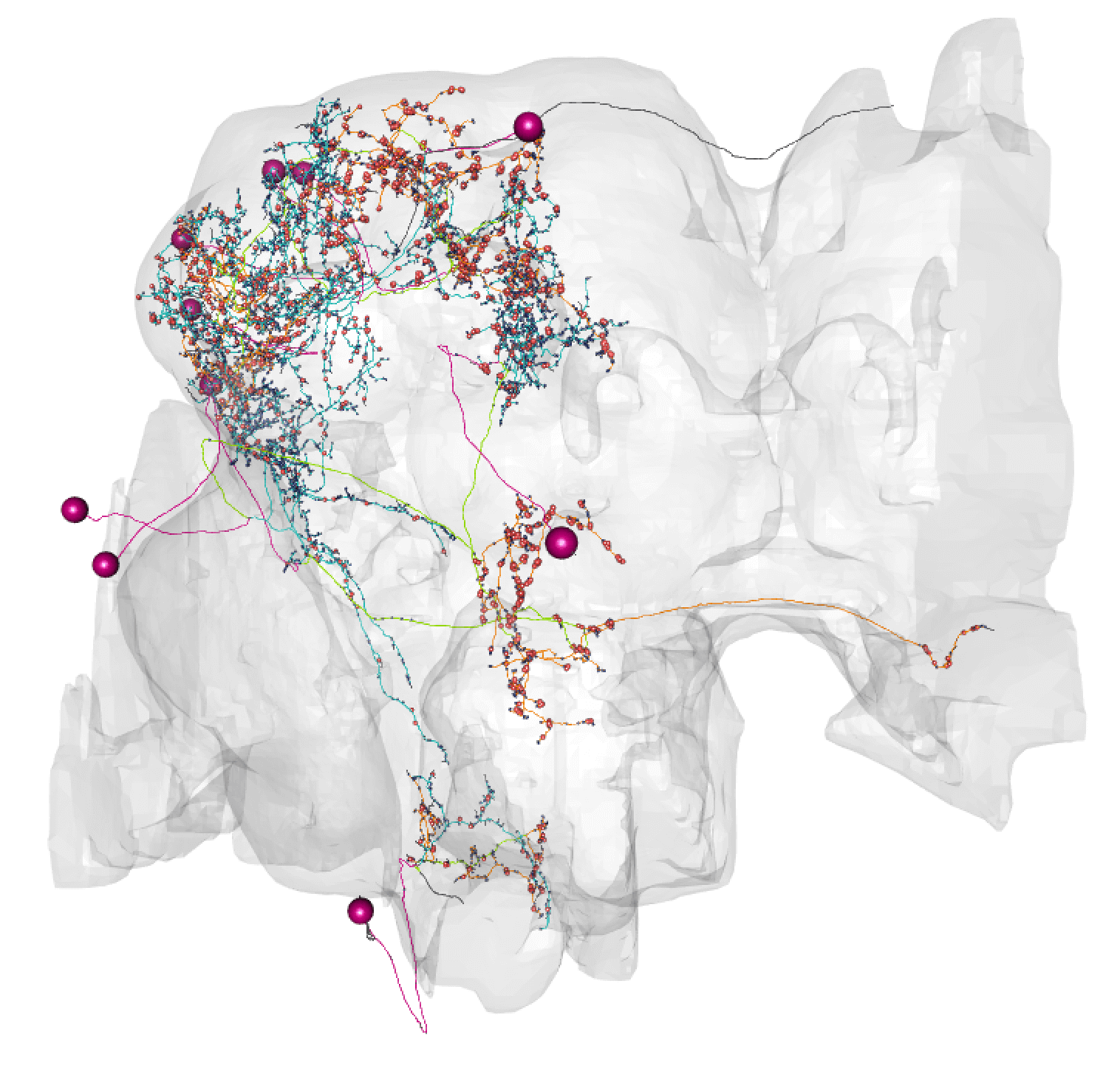
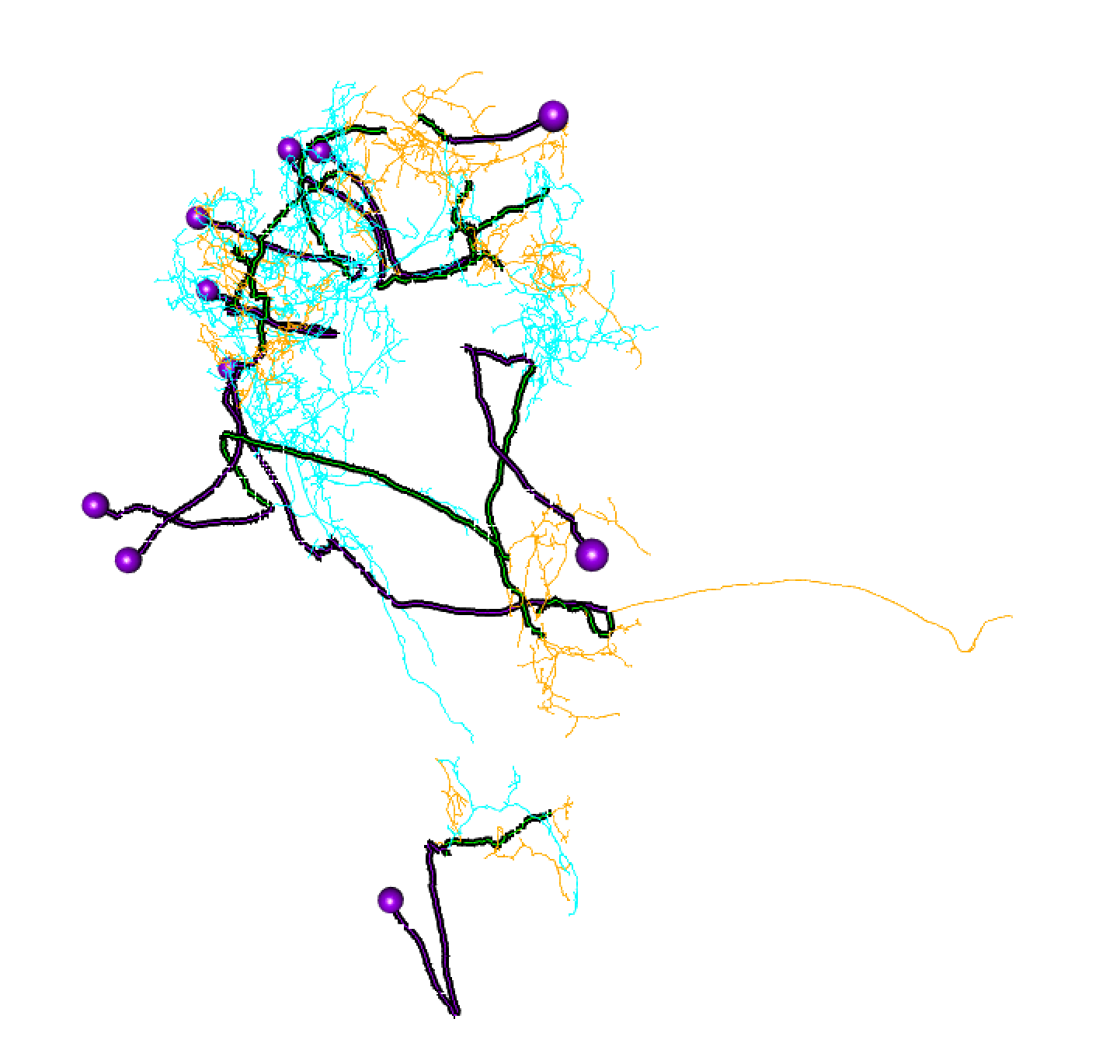
Here, lines: blue = dendrite, orange = axon, green = linker, purple = cell body fibre.
Balls: red = presynapse (output), navy blue = postsynapse (input), purple = soma.
@@ -258,7 +261,7 @@clear3d() nlscan_split(neurons.flow)
- 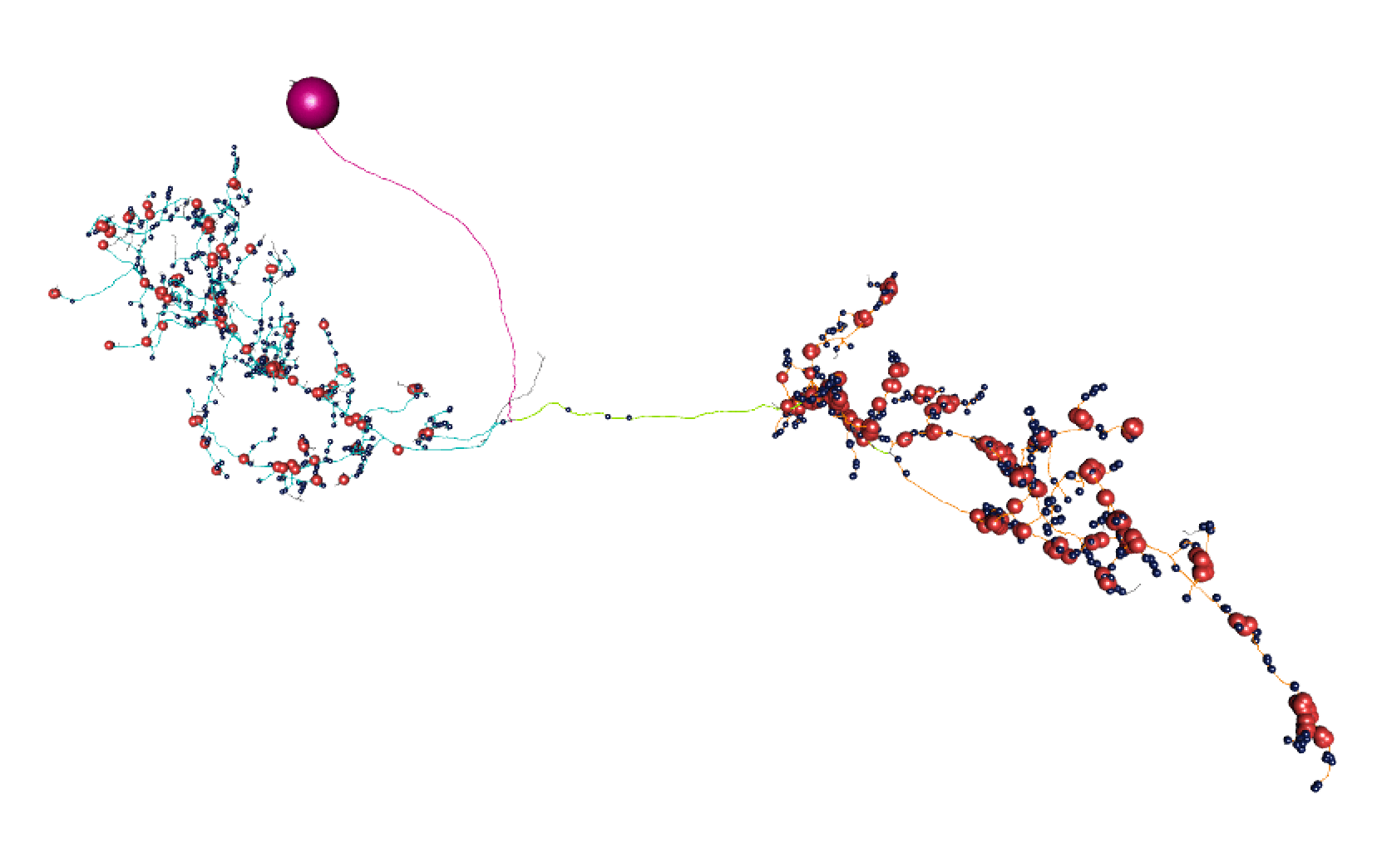 +
+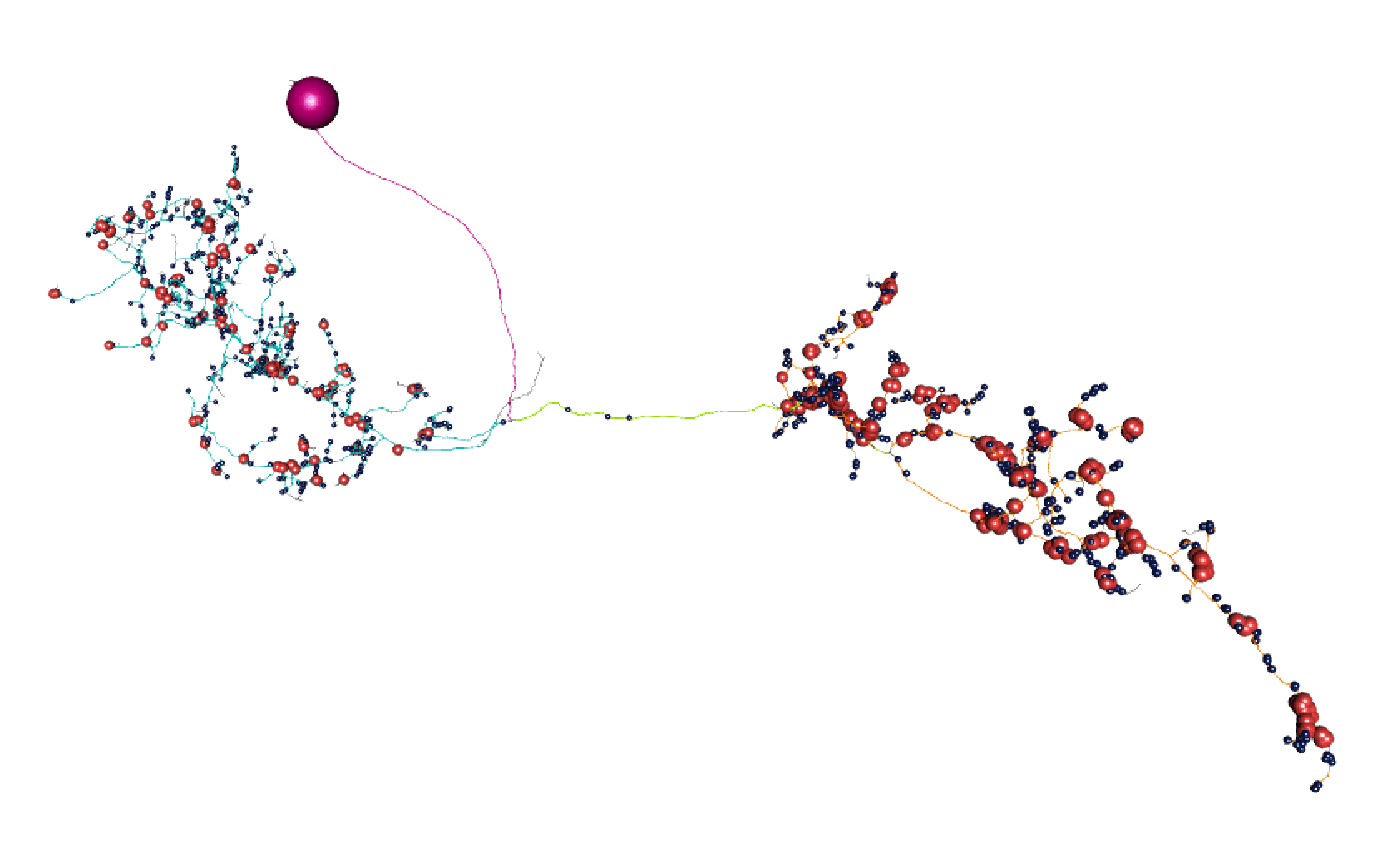
So I think most of those splits are pretty convincing.
@@ -310,7 +313,7 @@sort(table(pns.elist.strong$connection)) ## A lot of axo-axonic and dendro-dendritic action amongst the PNs
Here, pre is the
-bodyidof the presynaptic (upstream source) neuron and post is thebodyidof the postsynaptic (downstream target) neuron. Thenormcolumn gives a synaptic weight normalised by the total inputs of the downstream neuron (post), i.e. count/total postsynapses on ‘post’.Please see this article to see a more in-depth example of how to efficiently work neuron connectivity data from
+hemibrainr.Please see this article to see a more in-depth example of how to efficiently work neuron connectivity data from
diff --git a/docs/articles/hemibrain_connectivity.html b/docs/articles/hemibrain_connectivity.html index 279b40d9..8912b974 100644 --- a/docs/articles/hemibrain_connectivity.html +++ b/docs/articles/hemibrain_connectivity.html @@ -55,6 +55,9 @@hemibrainr.- + hemibrain and flywire meta data + +
- flywire
-
@@ -114,8 +117,8 @@
hemibrain connectivity
Connection data
-In this guide, we will use
-hemibrainrlook at connectivity data for a starting neuron of interest. They neuron will be LHMB1 (Bates & Schlegel et al., 2020)), the only neuron from the ‘innate olfactory’ brain center of the fly (lateral horn), to innervate the ‘associative memroy centre’ directly (mushroom body lobes). You can try re-running this analysis for any neuron of your interest.We will use precomputed data from the
+hemibrainrGoogle team drive. In order to connect R to this Google drive, you have a few options. Please see this article. To see how to split neurons into axon and dendrite, without acces to this drive, please see this article.In this guide, we will use
+hemibrainrlook at connectivity data for a starting neuron of interest. They neuron will be LHMB1 (Bates & Schlegel et al., 2020)), the only neuron from the ‘innate olfactory’ brain center of the fly (lateral horn), to innervate the ‘associative memory centre’ directly (mushroom body lobes). You can try re-running this analysis for any neuron of your interest.We will use precomputed data from the
hemibrainrGoogle team drive. In order to connect R to this Google drive, you have a few options. Please see this article. To see how to split neurons into axon and dendrite, without access to this drive, please see this article.Load google drive
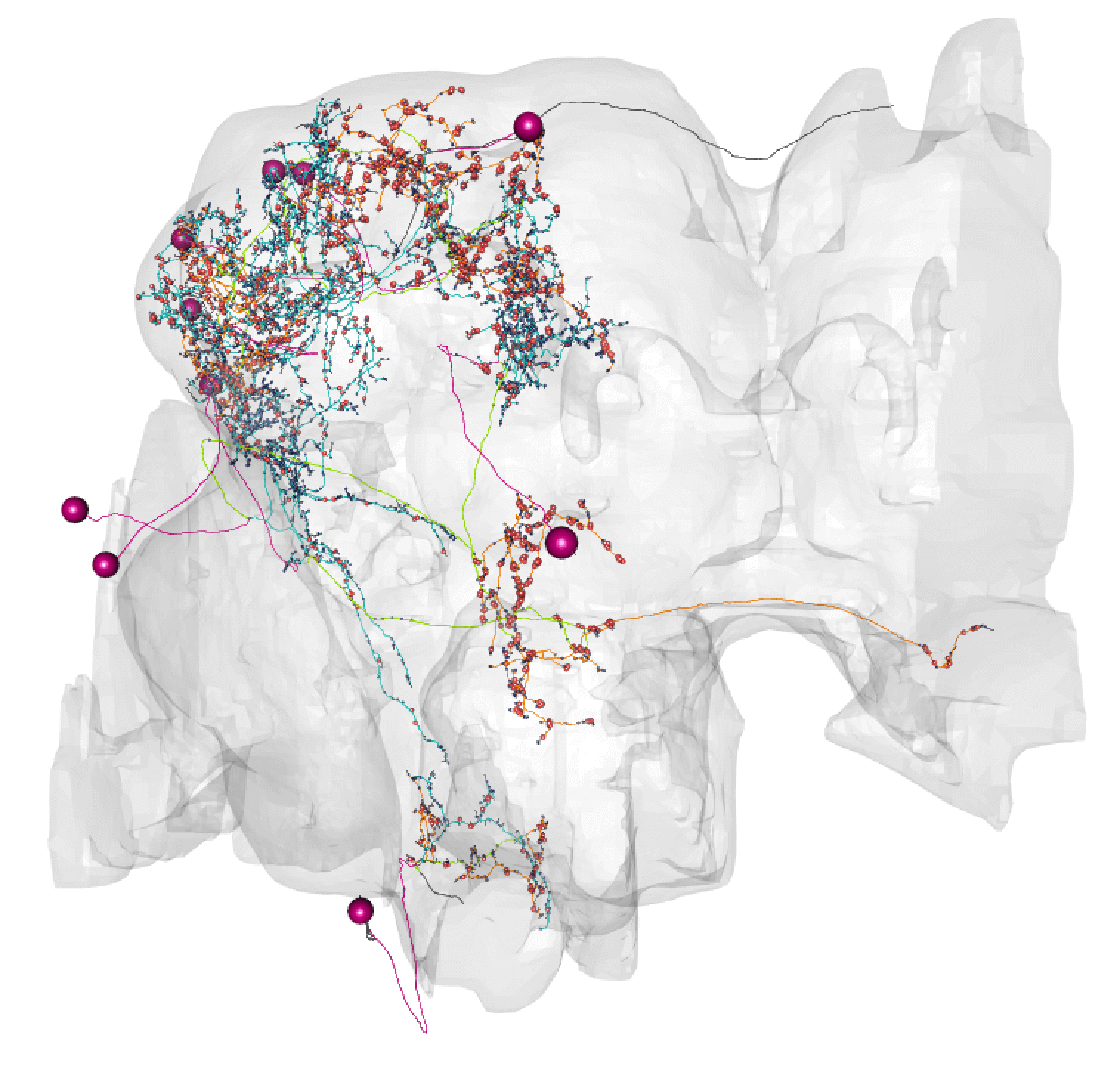
@@ -161,14 +164,14 @@plot3d(hemibrain.surf, alpha = 0.1, col = "lightgrey", add = TRUE) plot3d_split(lhmb1.neuron, lwd =2, soma = 600)
- -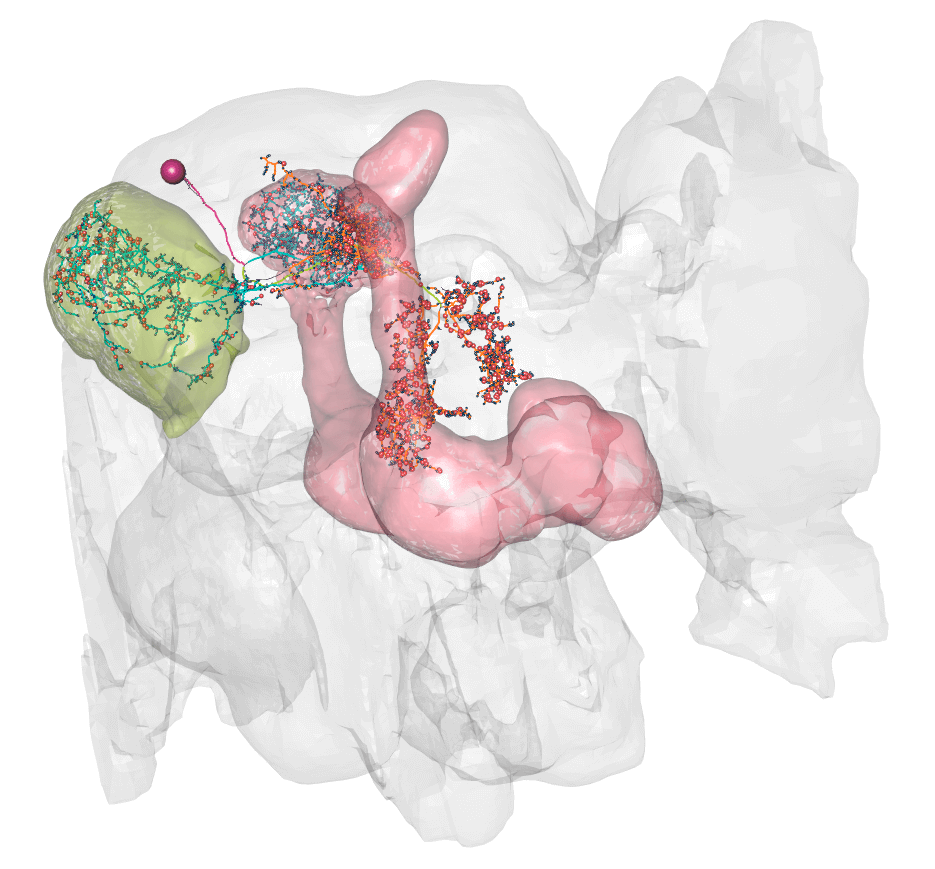 +
+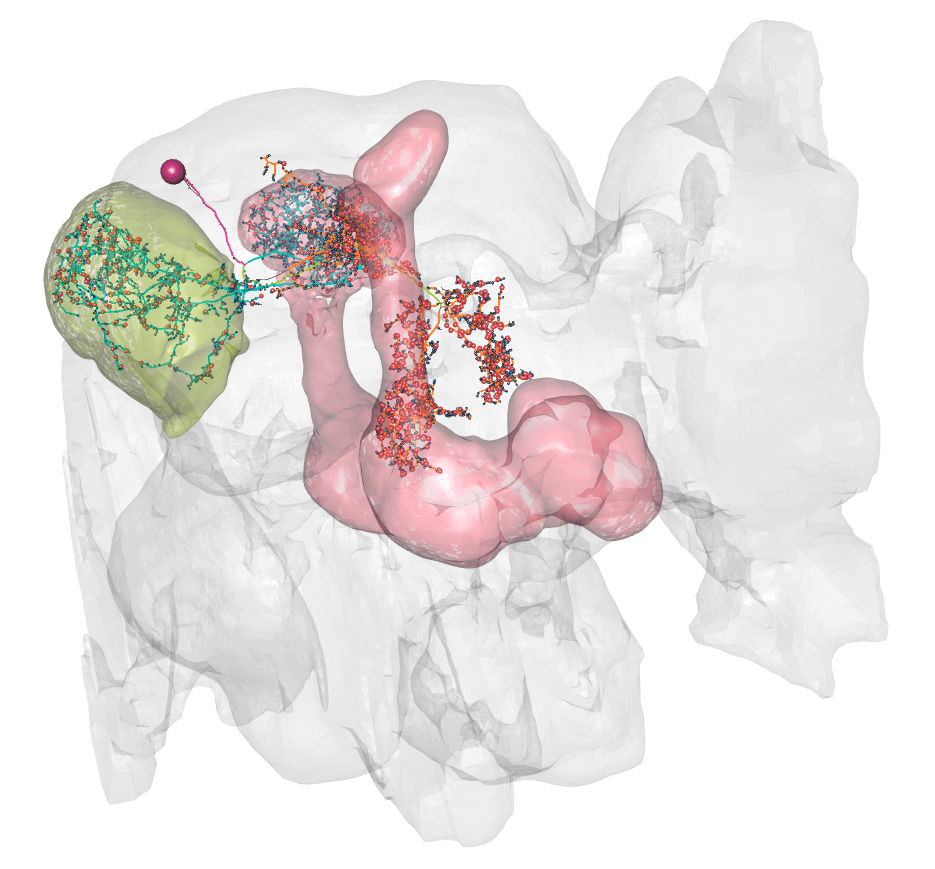
So here you can see dendrite (blue) in the lateral horn (green volume) and calyx, and axon (orange) in the lobes of the msuhroom body (pink volume).
+So here you can see dendrite (blue) in the lateral horn (green volume) and calyx, and axon (orange) in the lobes of the mushroom body (pink volume).
Get neuron connectivity data
-Now we can get a precomputed edgelist for the brain from the Google drive, which breaks down connections by axon and dendrite. By default this is read from an SQLlite database on the Google drive. This means we can access the data, without loading a multi Gb .csv into memory:
+Now we can get a precomputed edgelist for the brain from the Google drive, which breaks down connections by axon and dendrite. By default this is read from an SQLite database on the Google drive. This means we can access the data, without loading a multi Gb .csv into memory:
-# Get the edgelist elist = hemibrain_elist() @@ -178,8 +181,8 @@# Get a lot of meyta data for each hemibrain neuron hemi.meta = hemibrain_meta()
In our
-elist, count is the number of synaptic connections between two arbours. The arbour types are given byLabelandpartner.Label. The neuron identities are given aspre, the upstream (source) neuron andpost, the downstream (target) neuron. The value ofnormis generated by takingcountand dividing it by the total number of posynapses on the downstream target’s given arbour (note, not all connection in the neuron!). See your article on neuron splitting, for more information.We have the connectivity of whe whole dataset at our fingertips. Or at least, all the large fragments that could be considered neurons (see
+?hemibrain_bodyids). Let us have a quick look at what rthe distribution of different connection types is:In our
+elist, count is the number of synaptic connections between two arbours. The arbour types are given byLabelandpartner.Label. The neuron identities are given aspre, the upstream (source) neuron andpost, the downstream (target) neuron. The value ofnormis generated by takingcountand dividing it by the total number of postsynapses on the downstream target’s given arbour (note, not all connection in the neuron!). See your article on neuron splitting, for more information.We have the connectivity of the whole dataset at our fingertips. Or at least, all the large fragments that could be considered neurons (see
?hemibrain_bodyids). Let us have a quick look at what the distribution of different connection types is:
@@ -204,12 +205,11 @@lhmb1.elist <- elist %>% dplyr::filter(pre == lhmb1.bodyid | post == lhmb1.bodyid, count > 10 | norm > 0.01) %>% diff --git a/docs/articles/index.html b/docs/articles/index.html index b99a6189..6af7131e 100644 --- a/docs/articles/index.html +++ b/docs/articles/index.html @@ -95,6 +95,9 @@-
+
- + hemibrain and flywire meta data +
- flywire @@ -147,6 +150,8 @@
- hemibrain and flywire meta data +
- flywire
- read data from Google drive diff --git a/docs/articles/match_making.html b/docs/articles/match_making.html index cf2ec7d0..aab3733d 100644 --- a/docs/articles/match_making.html +++ b/docs/articles/match_making.html @@ -55,6 +55,9 @@
- + hemibrain and flywire meta data + +
- flywire
-
@@ -116,42 +119,42 @@
Neuron Matching Making
Insect brains seem pretty stereotyped. But just how stereotyped are they? It comes as a surprise to many neuroscientists who work only on vertebrates, to learn that in insects, individual neurons can readily and reliably be re-found and identified across different members of the species. Perhaps even across species.
-  +
+
As of 2020, two large data sets for the vinegar fly, D. melanogaster, are available making it possible to look at the full morphology of ~25,000 neurons in two data sets. These data sets are the hemibrain and FAFB. However, neurons in FAFB have been semi-manually or manually reconstructed, making the automatic assignment of FAFB-hemibrain neuron matches non-trivial. In this R package we have built tools to enable users to record and deploy inter-dataset matches.
What use is this information? Matches could be used to look at morphological stereotypy, help find genetic lines that label neurons, help transfer information associated with on reconstructed to the same cell in a different brain, compare neuron connectivity between two brains, etc.
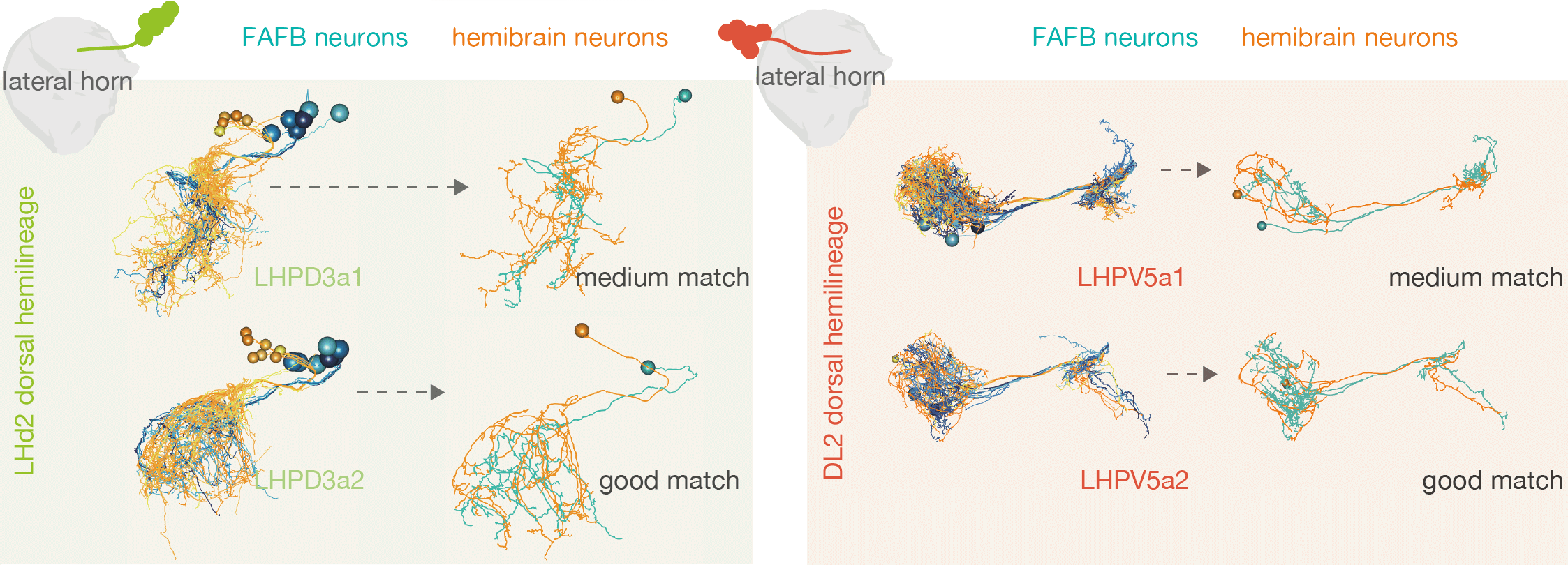
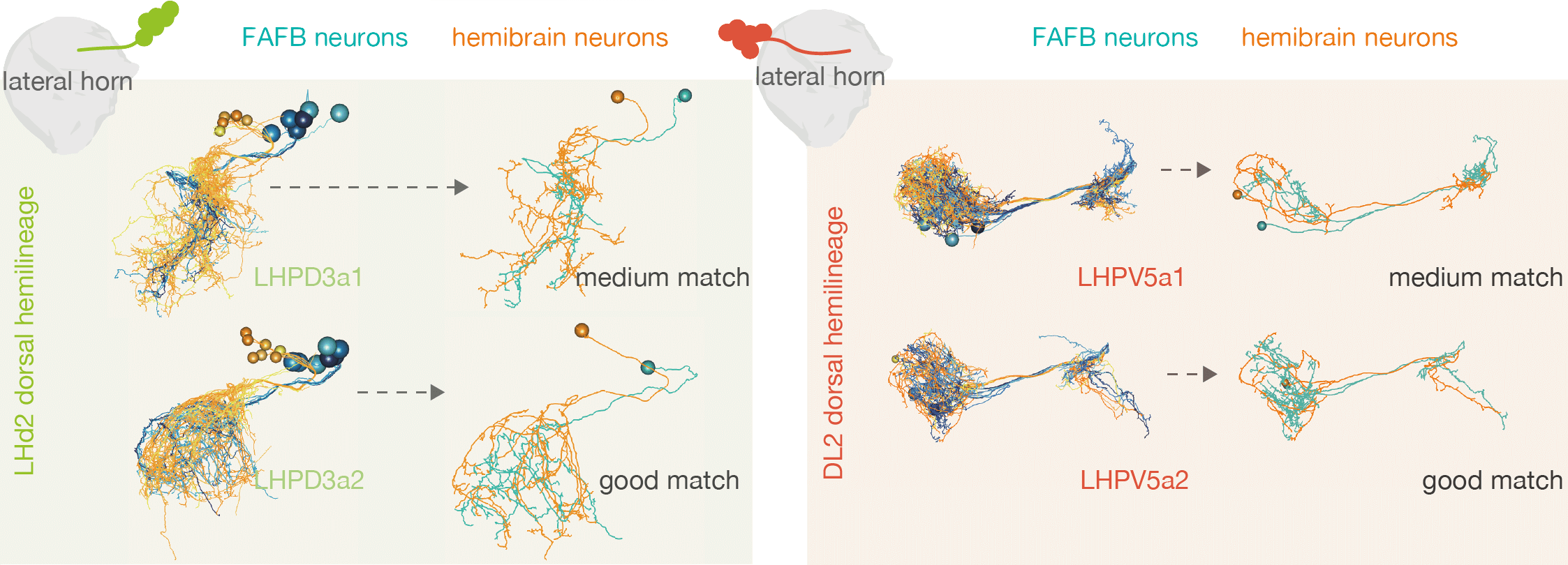
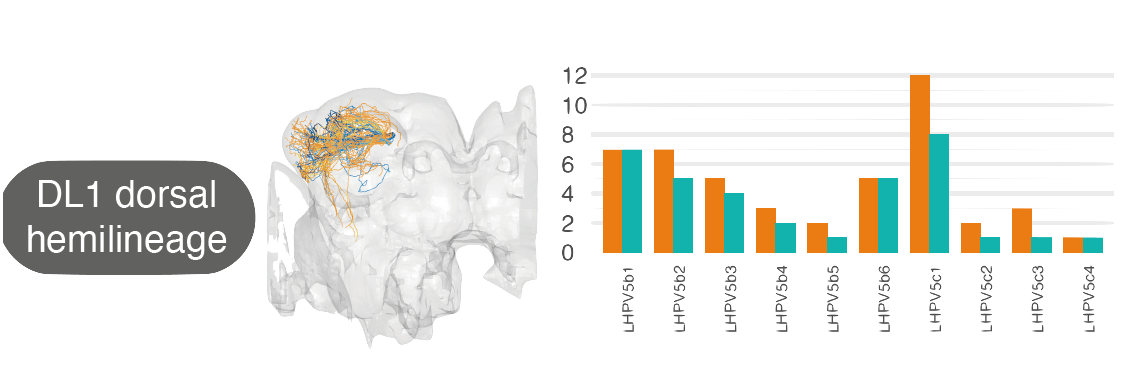
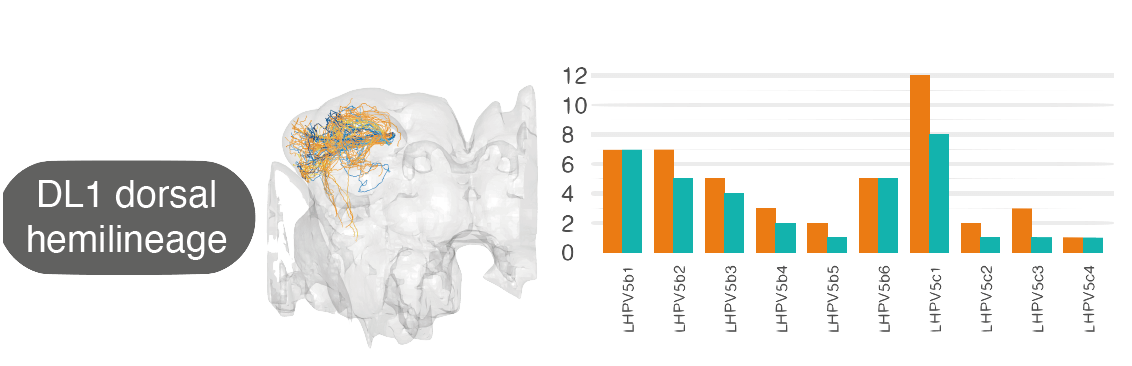
For example, by matching neurons up between the hemibrain and FAFB, we see that the numbers of cell types within one ‘hemilineage’ (a set of neurons that are born and develop together) are comparable between these two different flies:
-  +
+
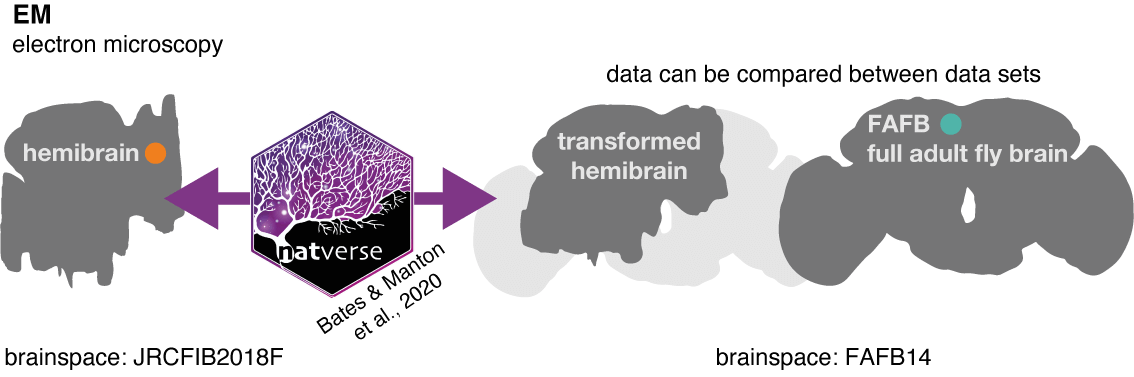
In order to match neurons, we make use of other natverse tools to ‘bridge’ data between two different brainspace, so they can be co-visualised (enabled by template brain and bridging registrations by Bogovic et al. 2019):
- 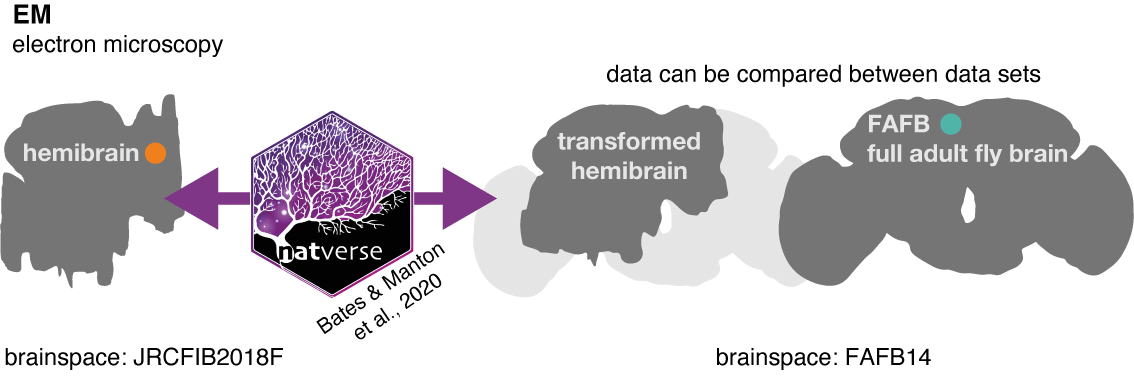 +
+
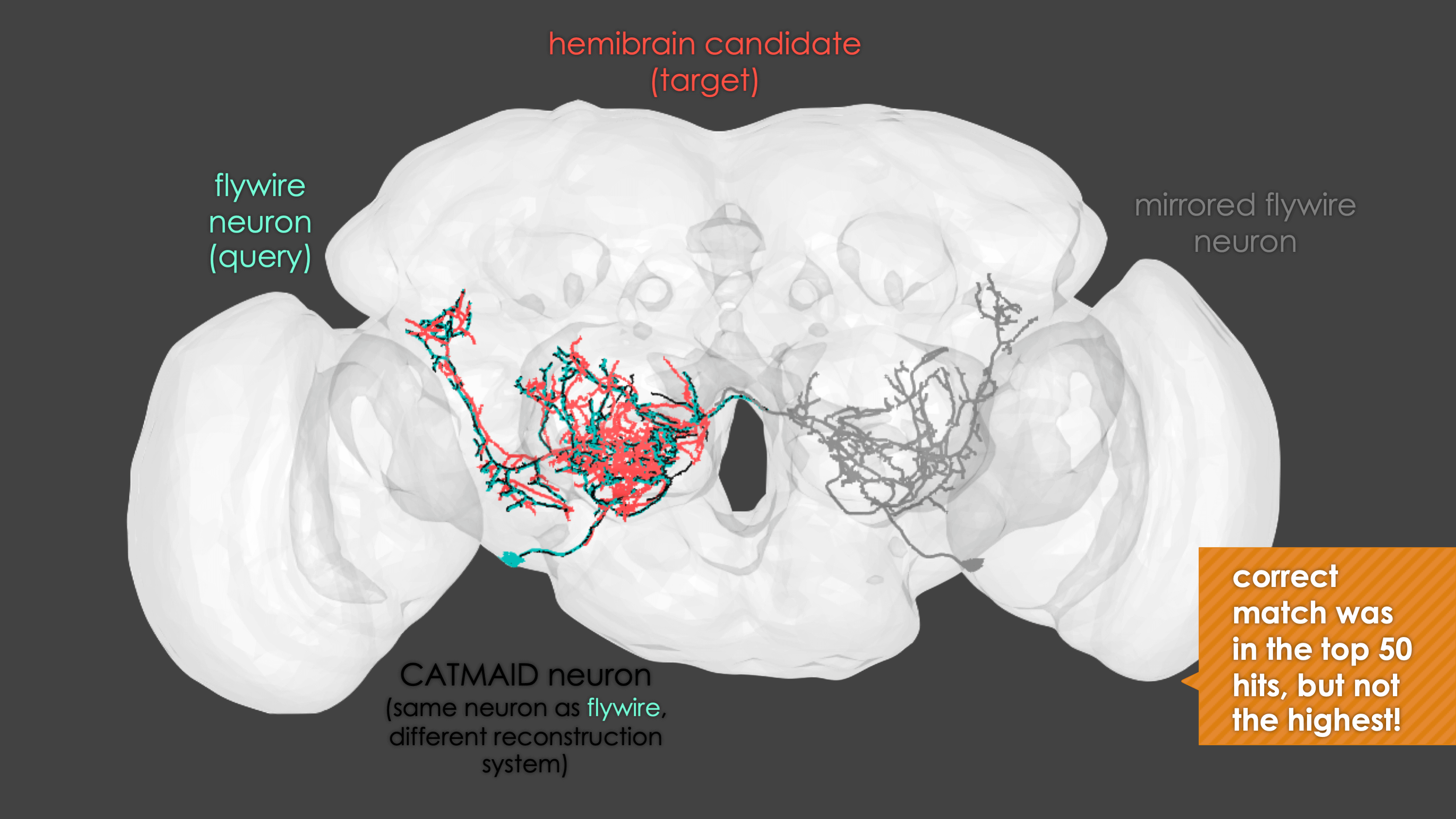
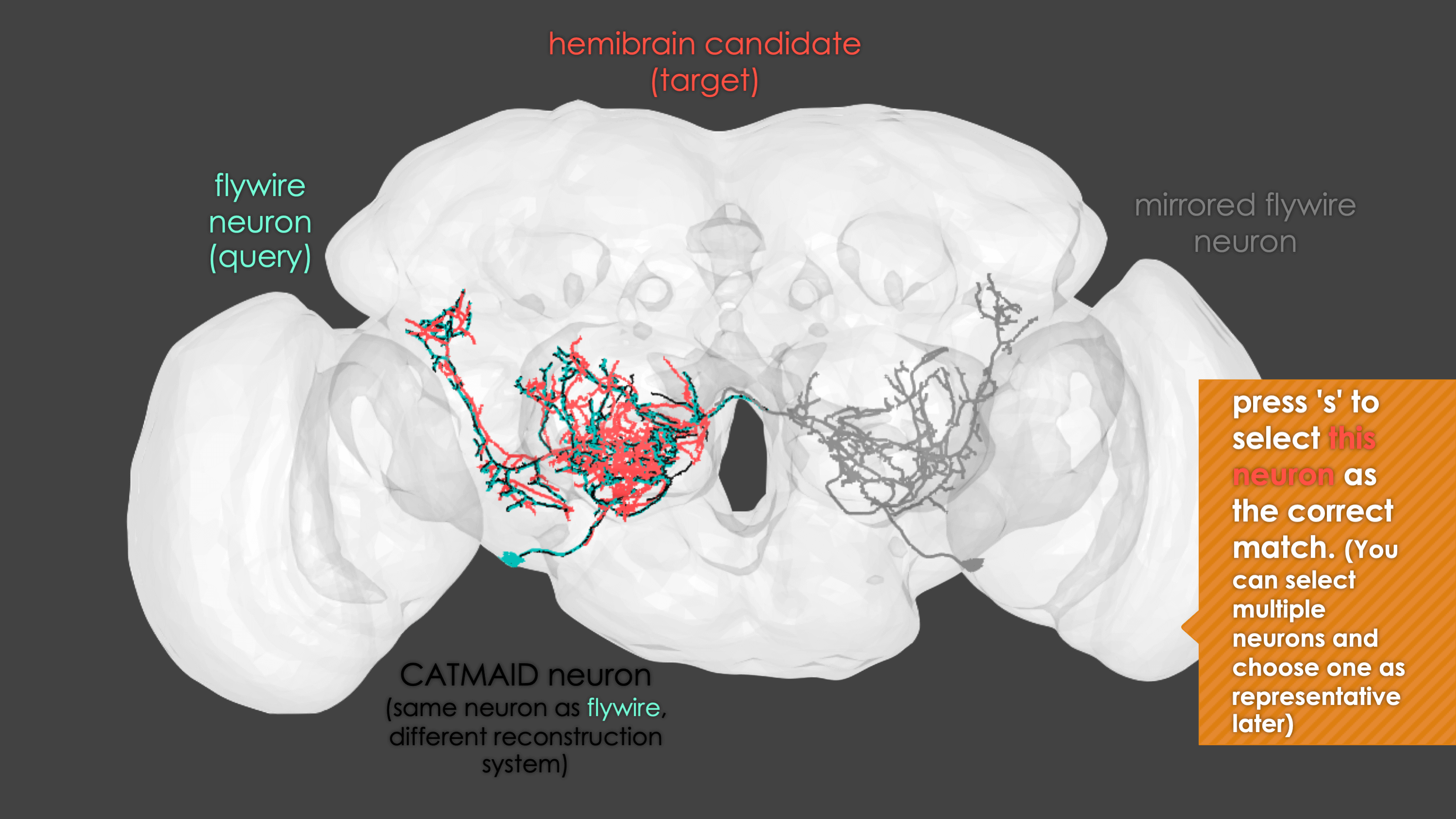
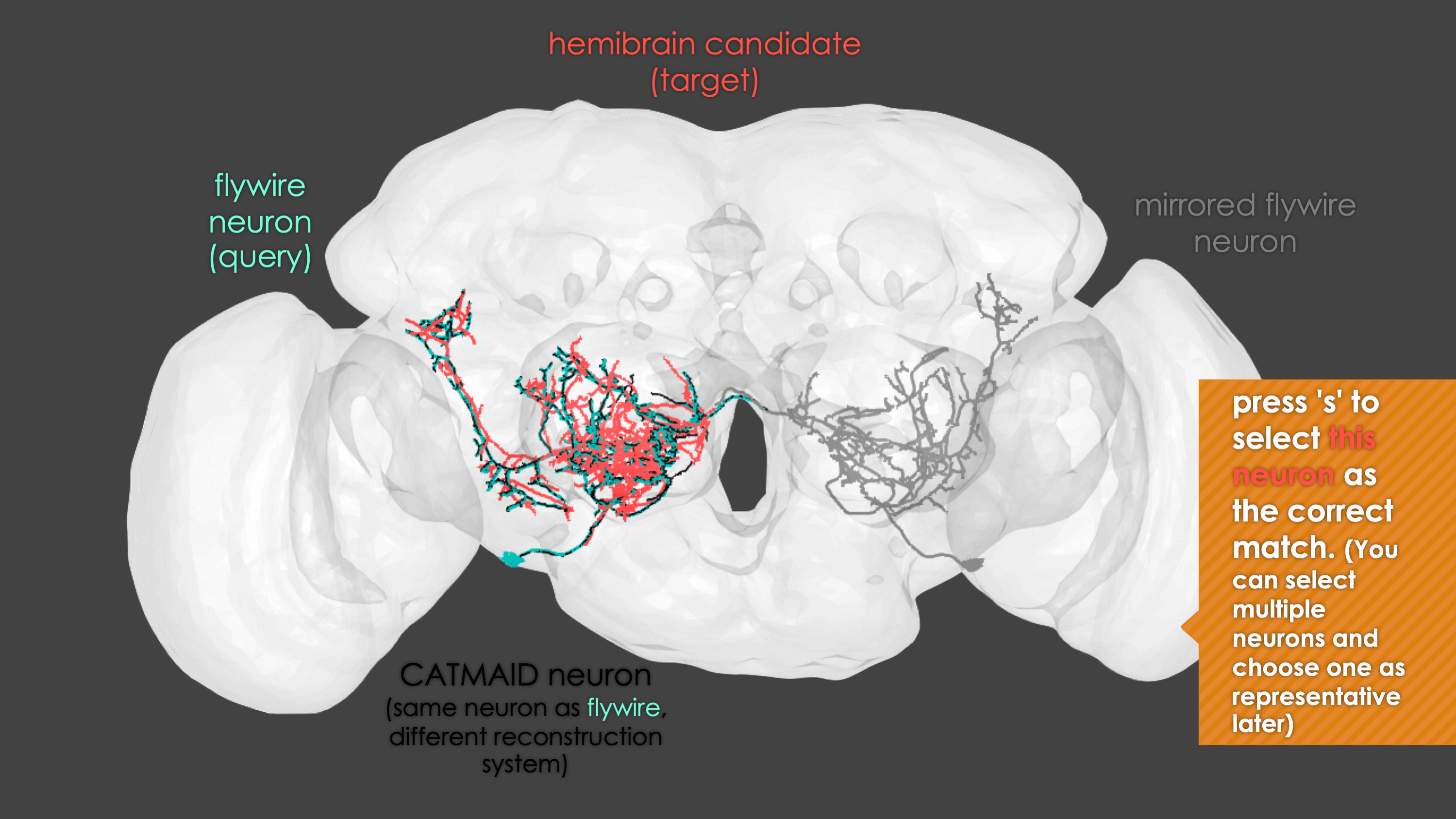
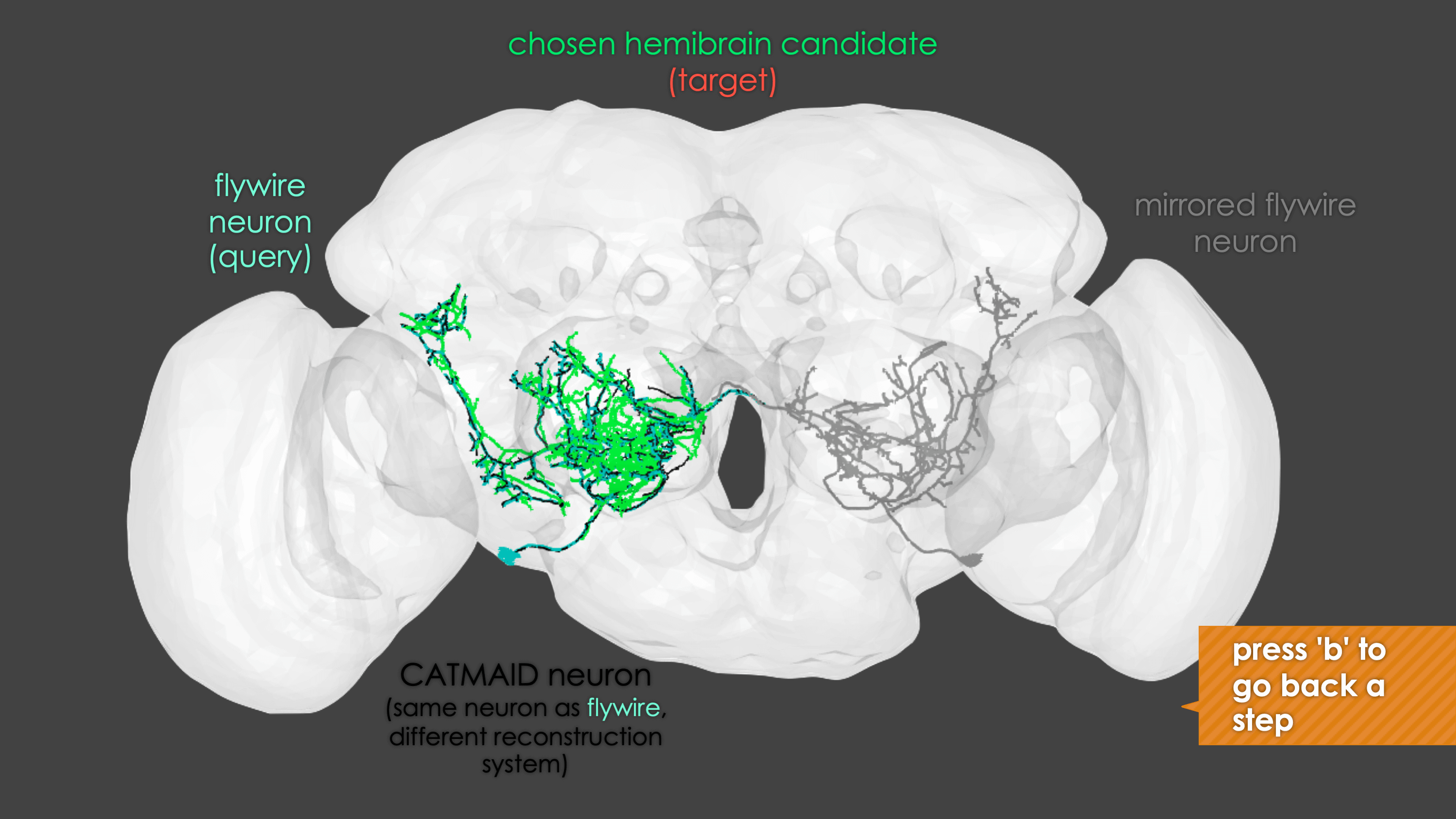
Overview
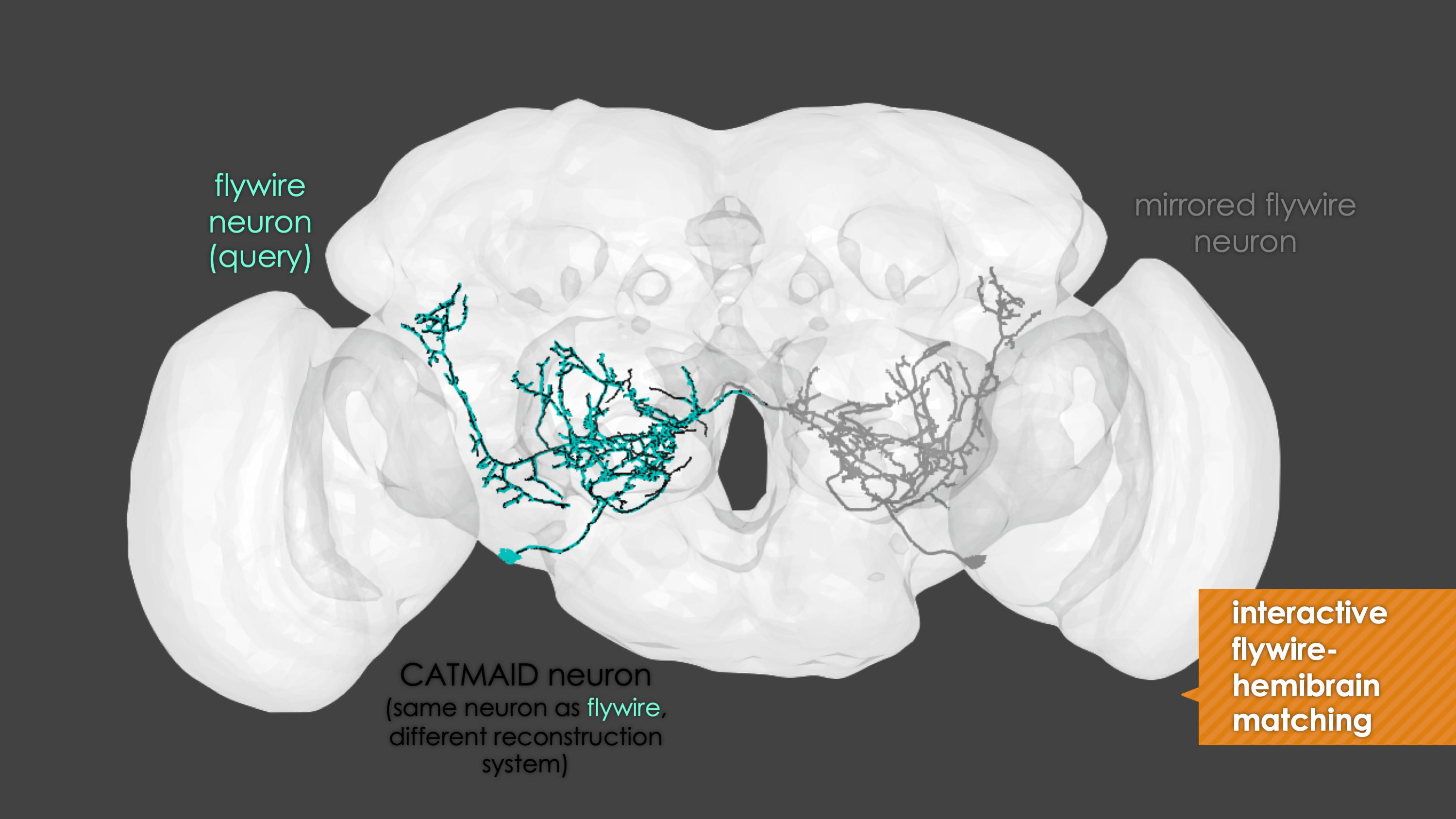
-In general, our interactive matching pipelines follow this workslow (this exmple is for
+fafb_matching):In general, our interactive matching pipelines follow this workflow (this example is for
fafb_matching):-  +
+
-  +
+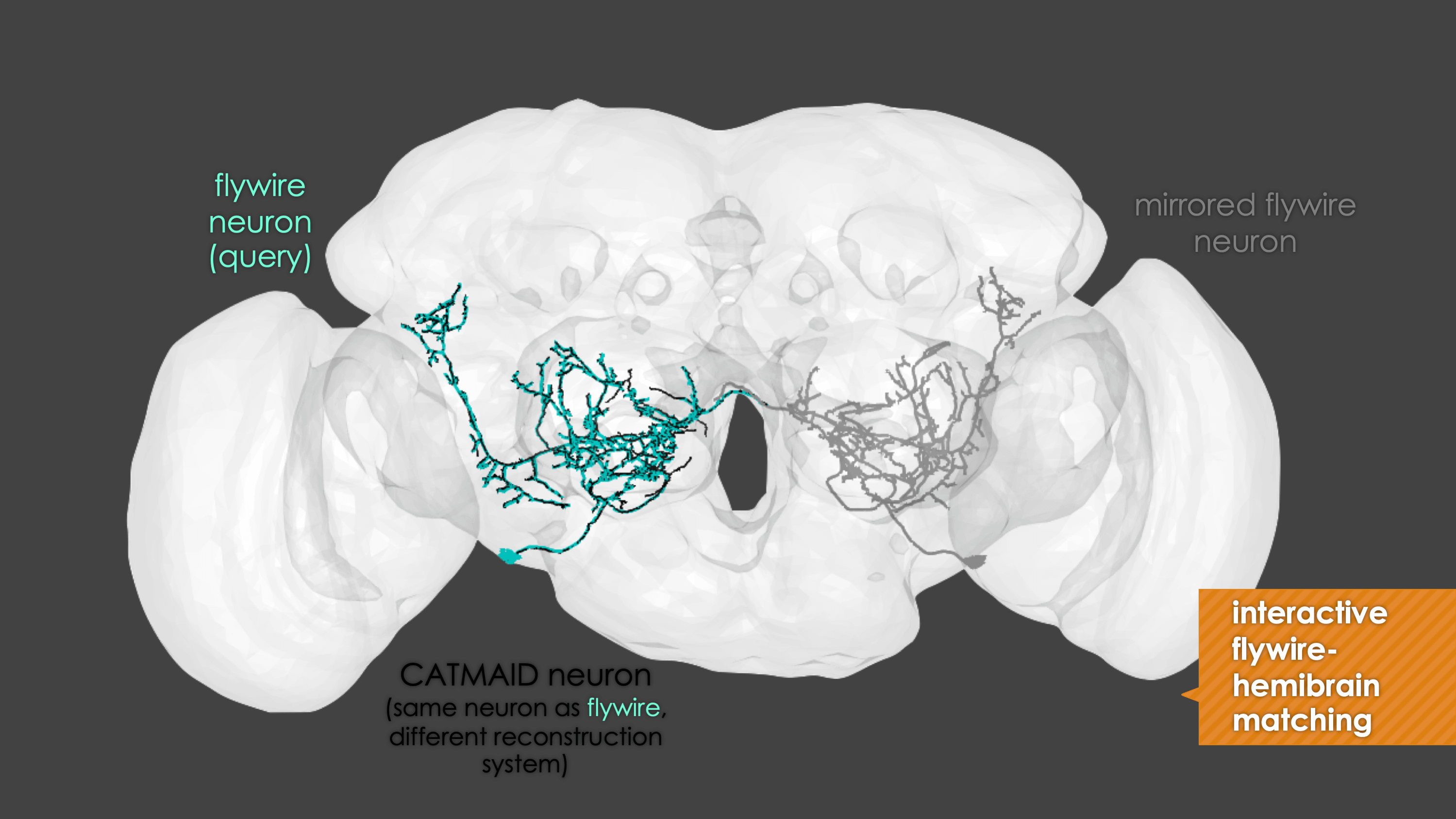
- 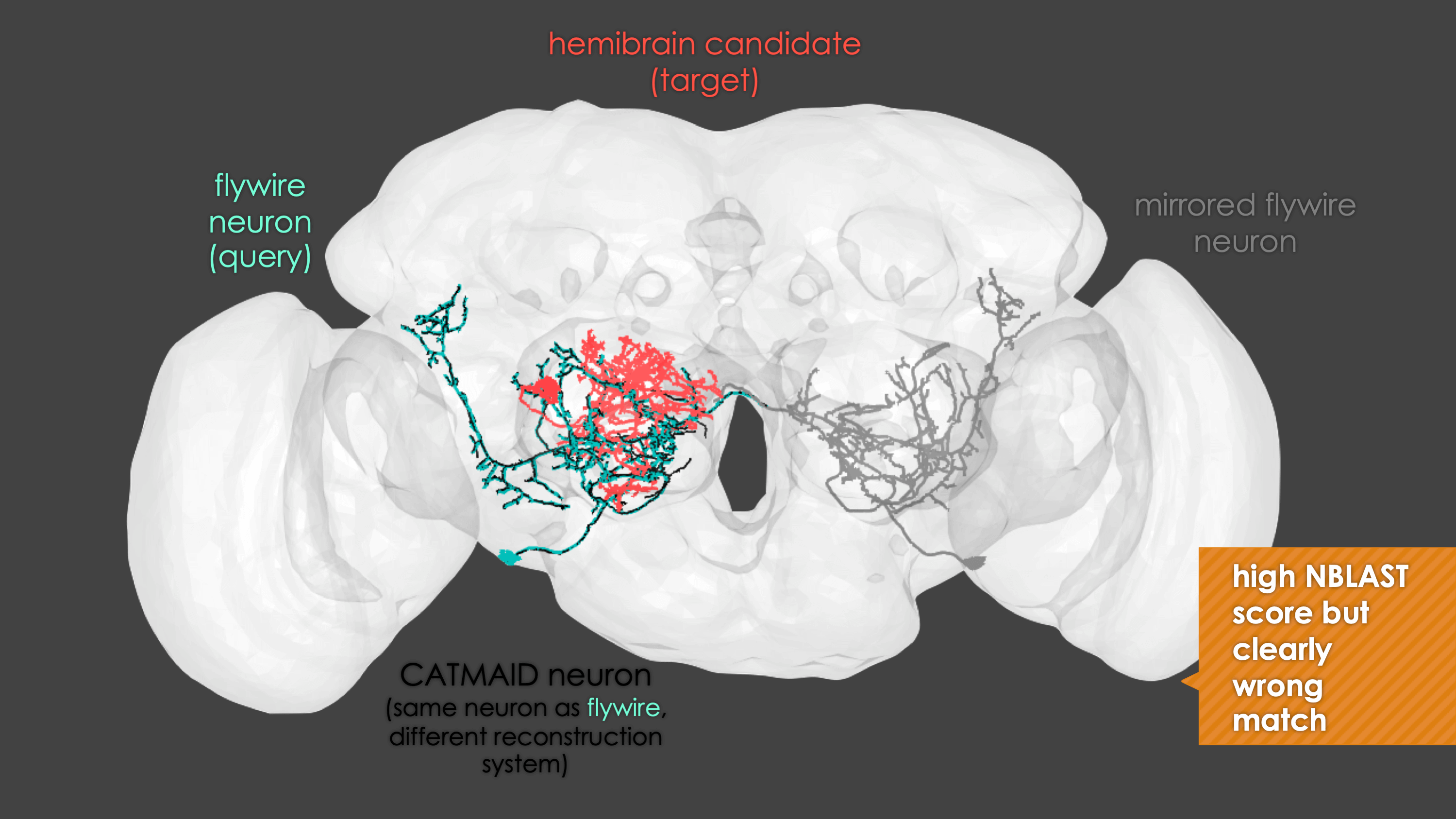 +
+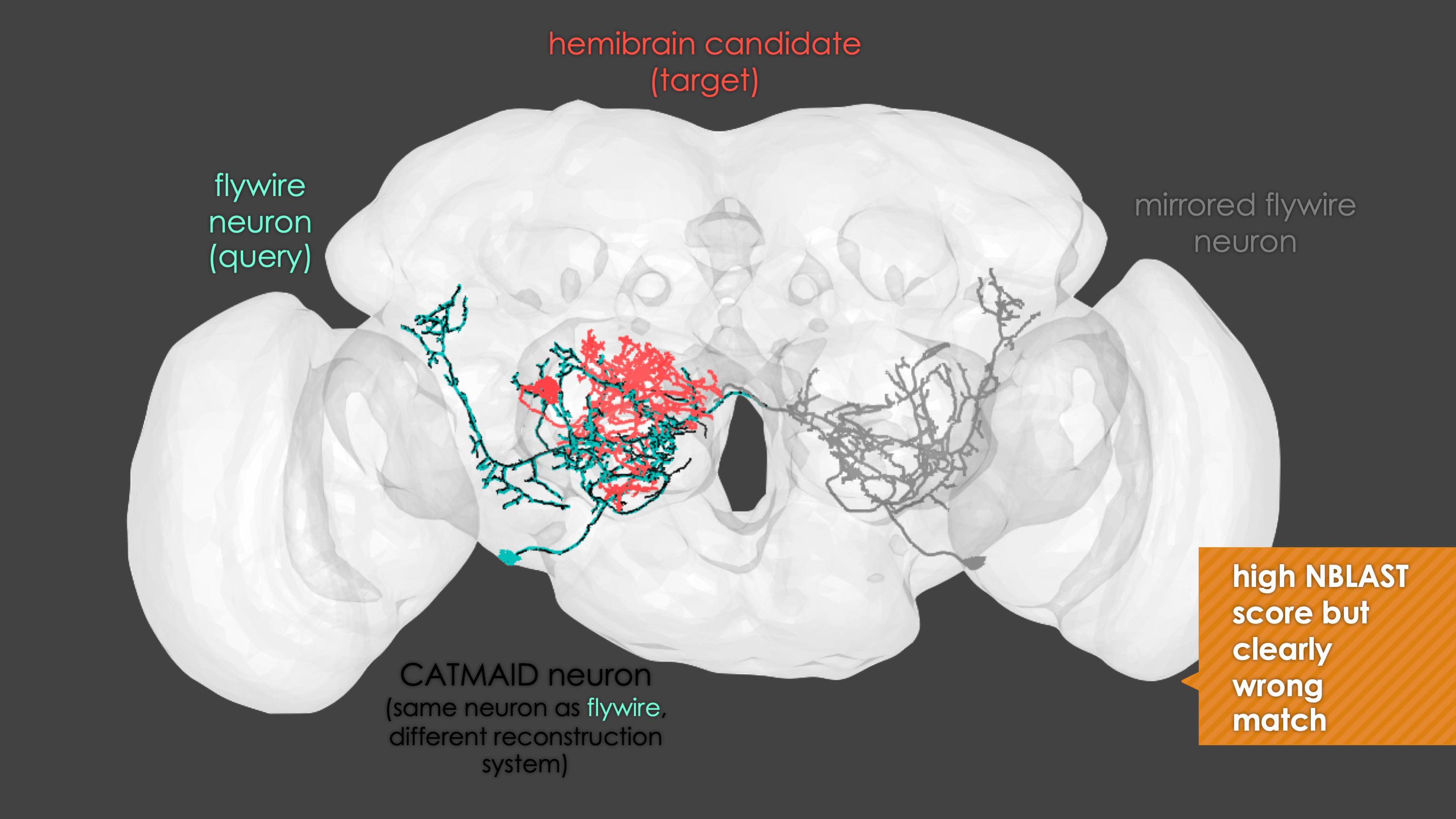
- 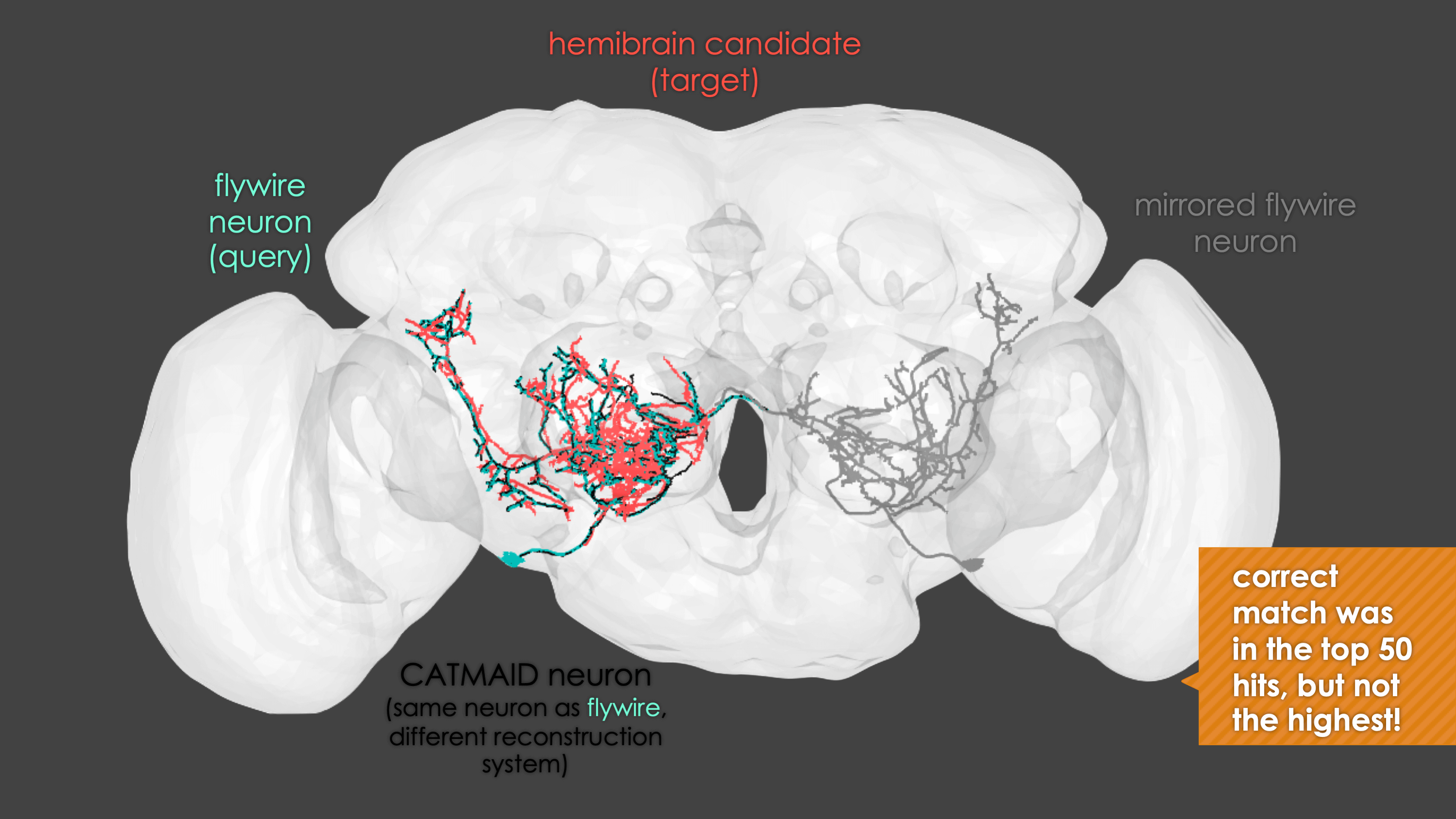 +
+
-  +
+
-  +
+
-  +
+ @@ -174,7 +177,7 @@diff --git a/docs/articles/packages_guide.html b/docs/articles/packages_guide.html index 23e9905c..10a81533 100644 --- a/docs/articles/packages_guide.html +++ b/docs/articles/packages_guide.html @@ -55,6 +55,9 @@
@@ -174,7 +177,7 @@diff --git a/docs/articles/packages_guide.html b/docs/articles/packages_guide.html index 23e9905c..10a81533 100644 --- a/docs/articles/packages_guide.html +++ b/docs/articles/packages_guide.html @@ -55,6 +55,9 @@The Google Sheet
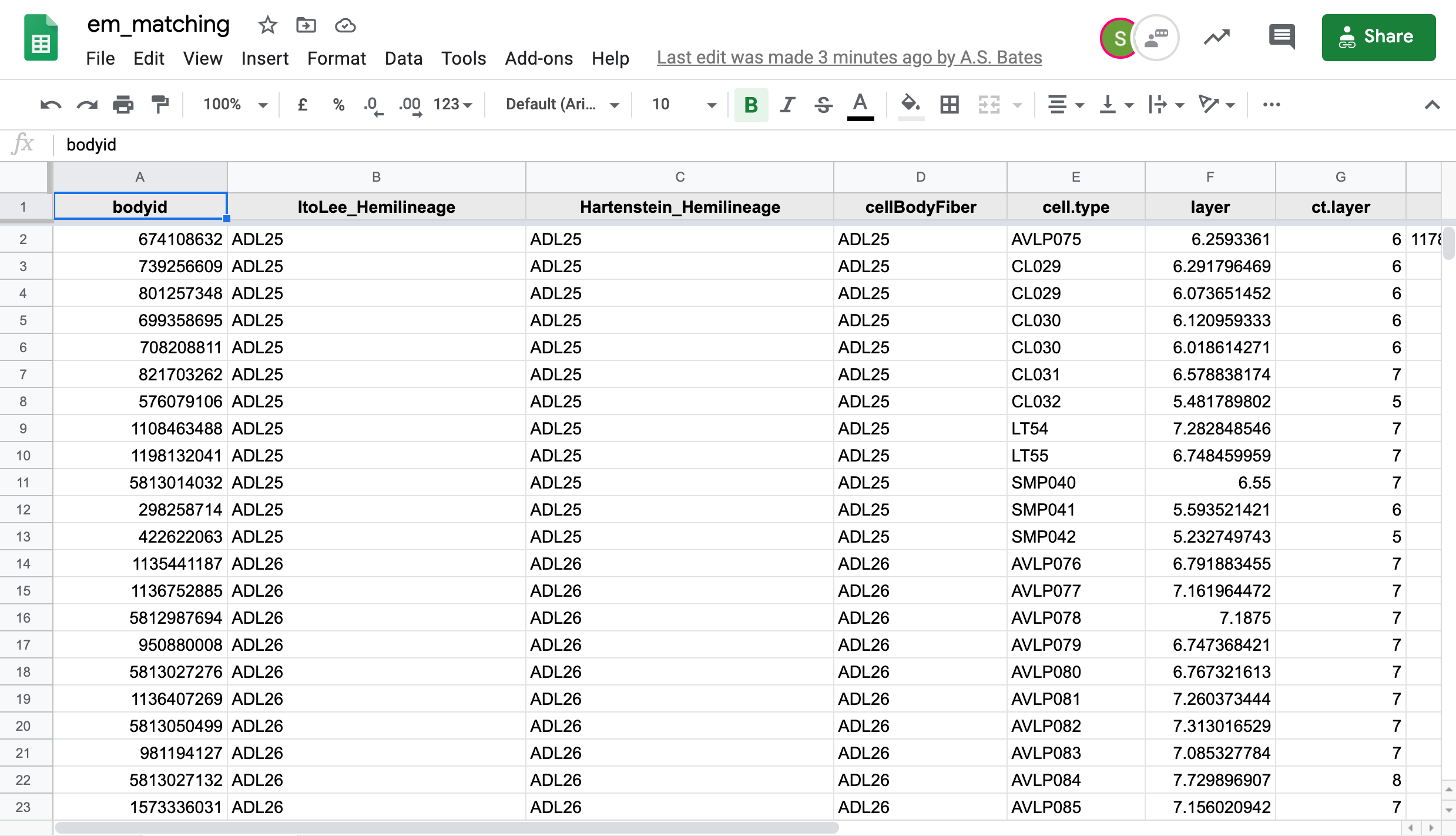
We in the Drosophila Connectomics Group have been recording our match making in a Google sheet named em_matching. This sheet has two tabs of concern here,
hemibrainfor hemibrain neuron -> FAFB neuron matches andfafbfor FAFB neuron to hemibrain neuron matches.- 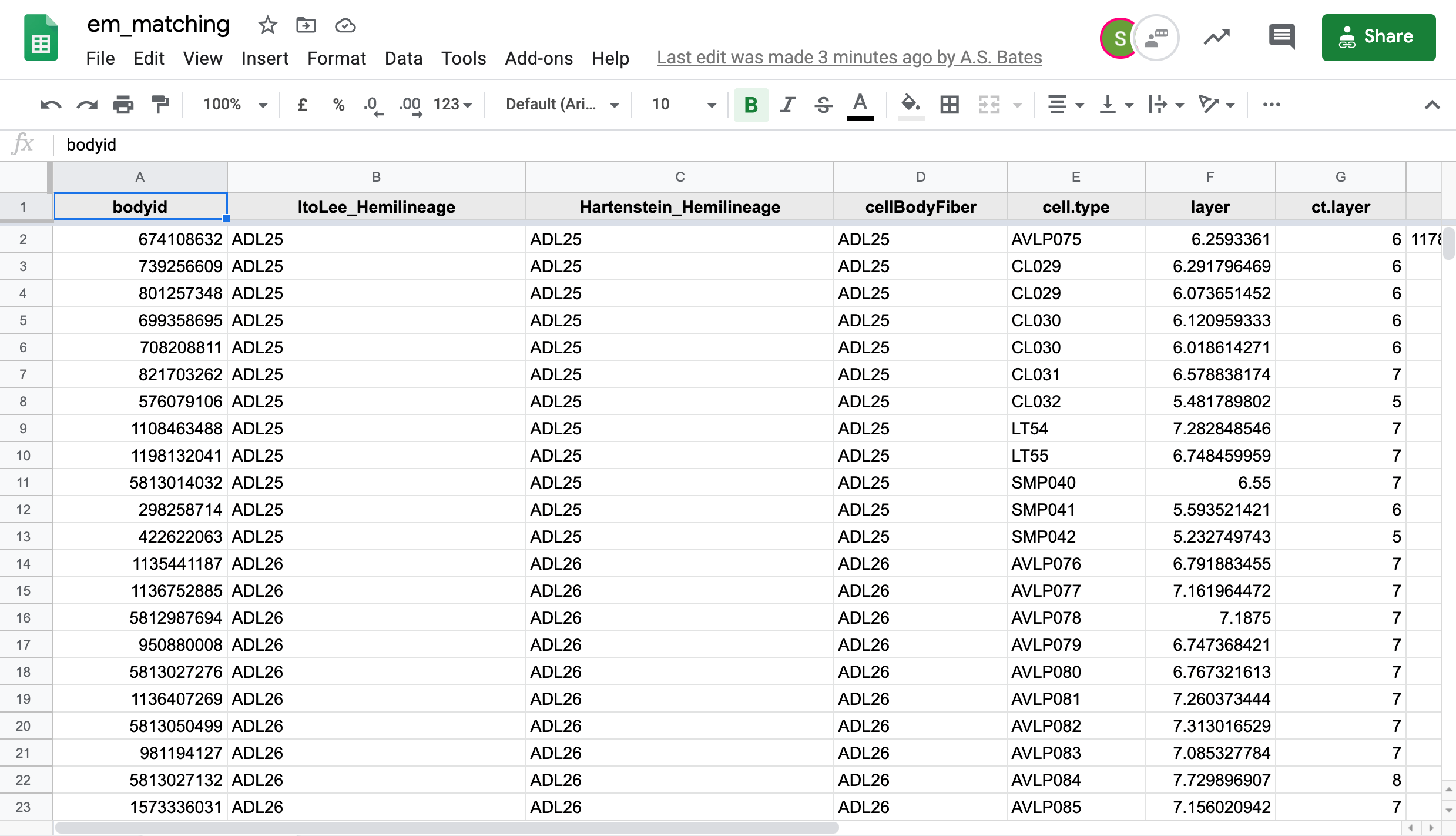 +
+
If you have authorisation, you can see the most up-to-date matches as so:
@@ -190,48 +193,48 @@
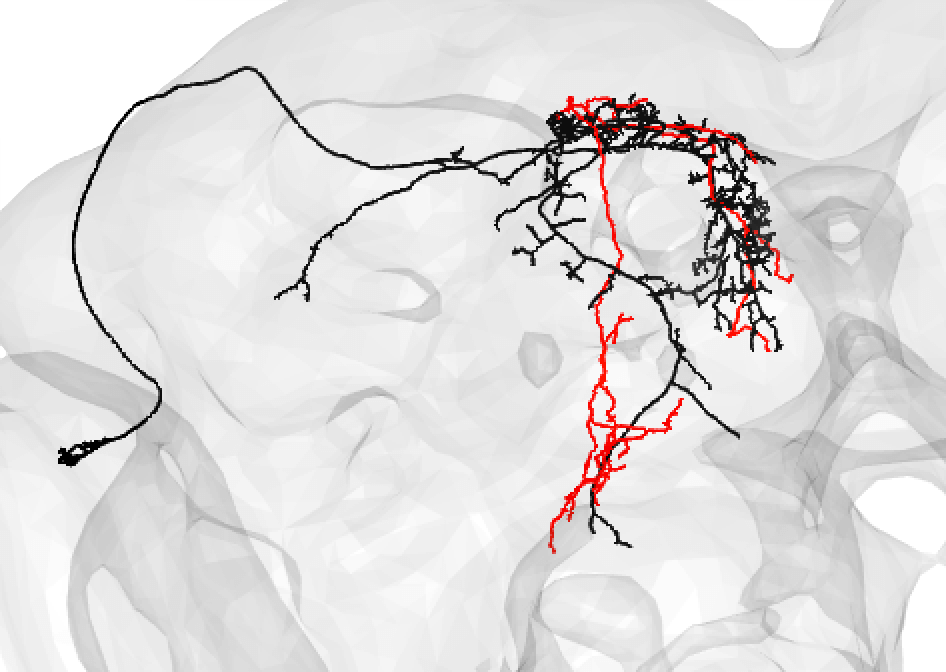
It is very important to note that a match cannot be a match if neurons do not seem to share the same cell body fiber tract. Being in a different tract is a deal breaker.
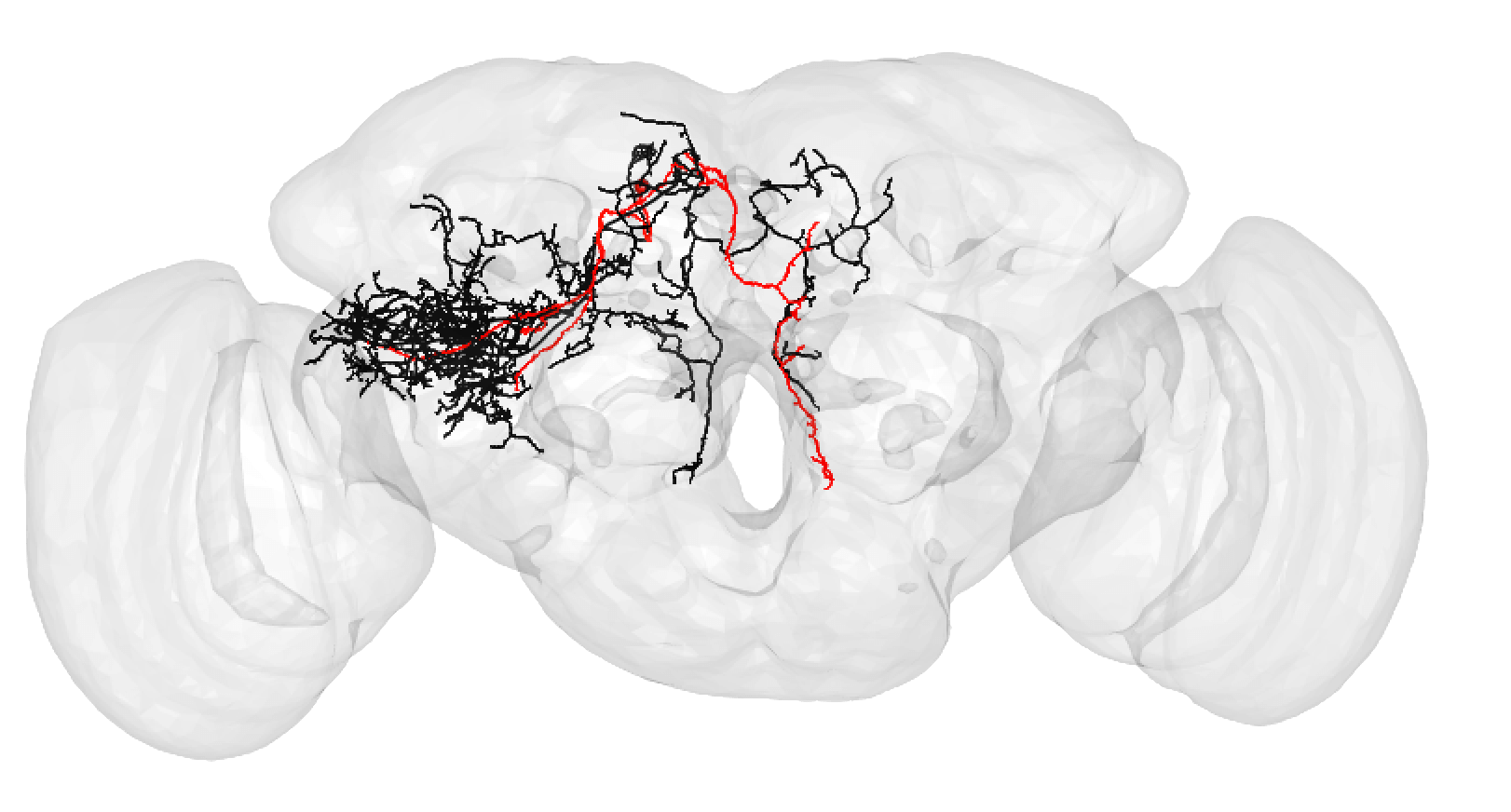
Some good matches are striking. For example:
- 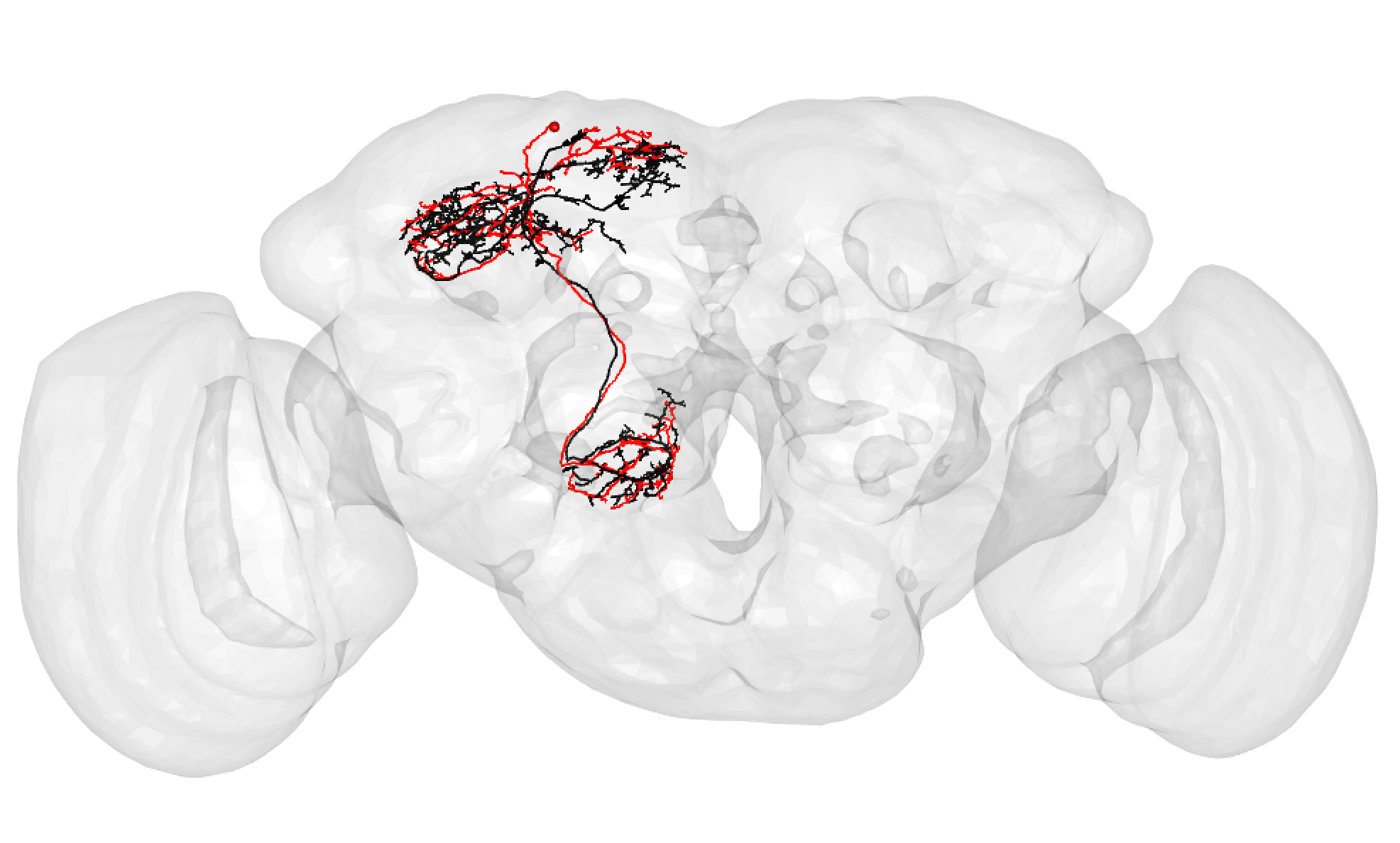 +
+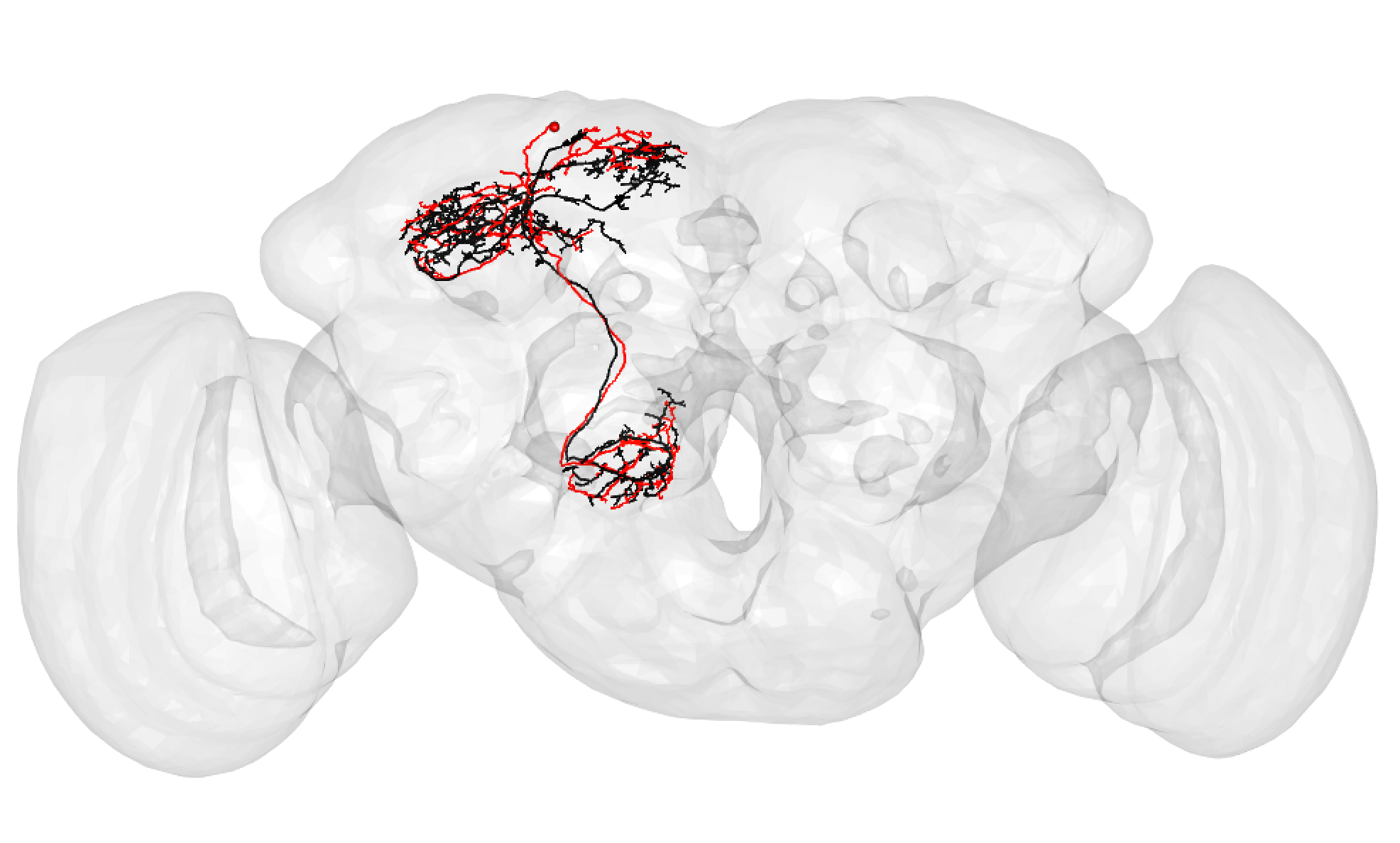
In the above case, the FAFB neuron has been quite extensively manually traced, meaning that these cells look very similar to one another.
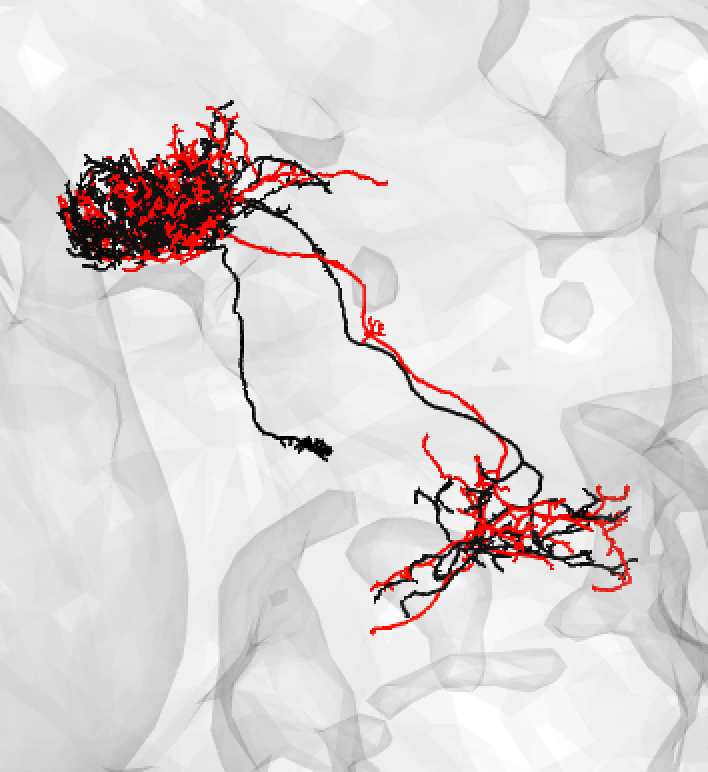
Be aware that while neurons must share the same cell body fiber tract, these tracts can be a little off set. For example, this is also a good match:
-  +
+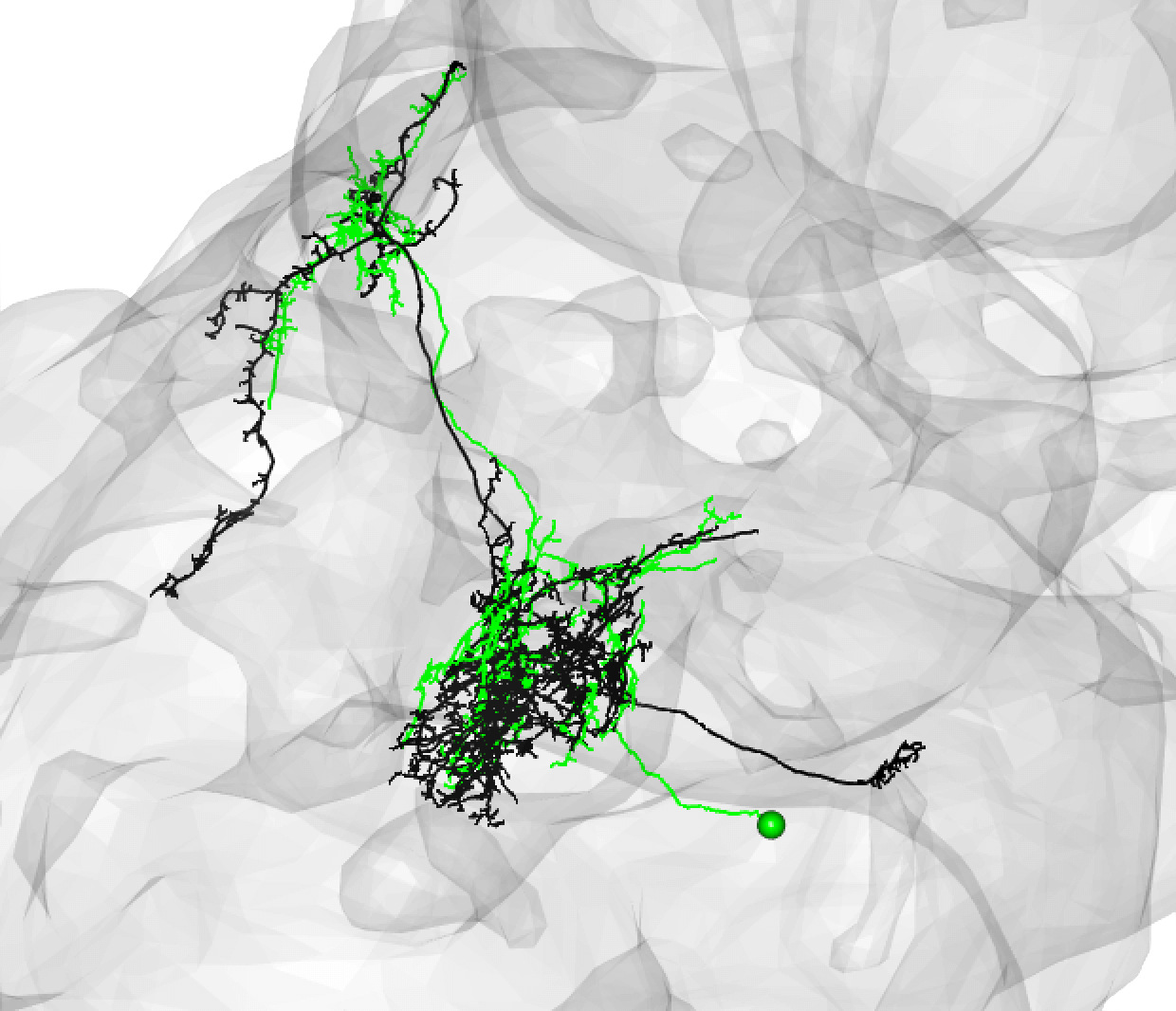
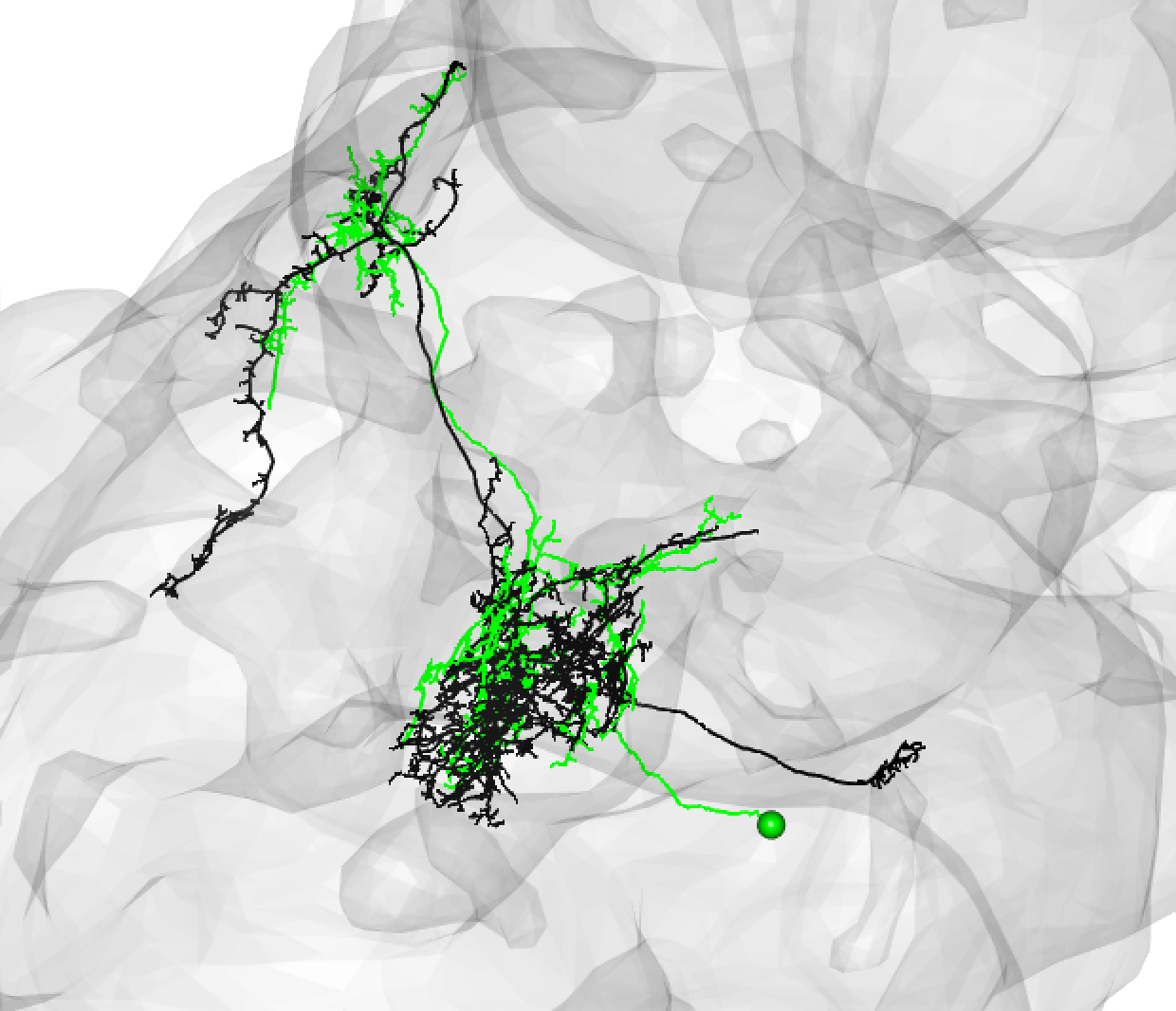
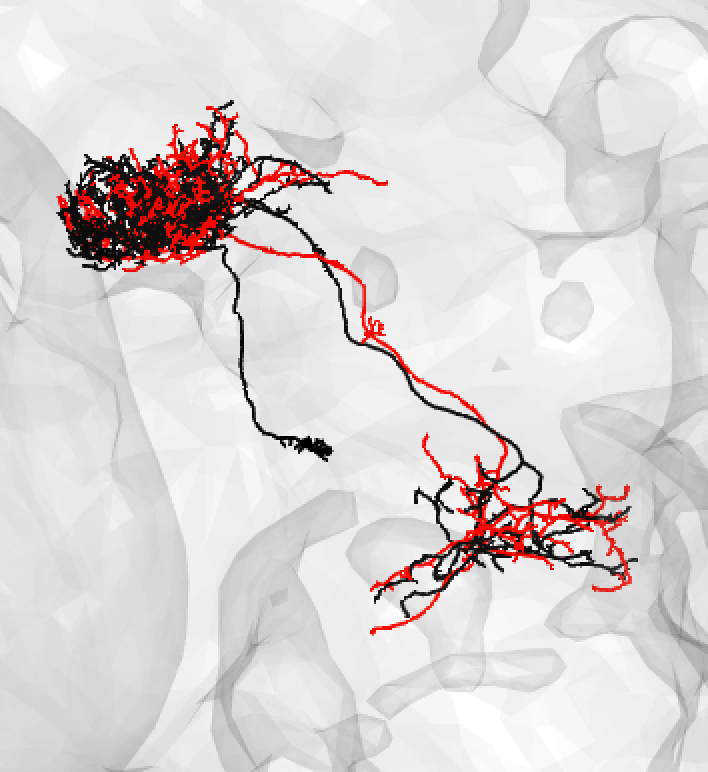
If the soma is missing, it might be safer to note a match as ‘medium’.
-  +
+
You might also use medium if you have a nice looking match and suspect that there is a medium/large discrepancy because the FAFB neuron (here shown in red) is under-traced, such as:
- 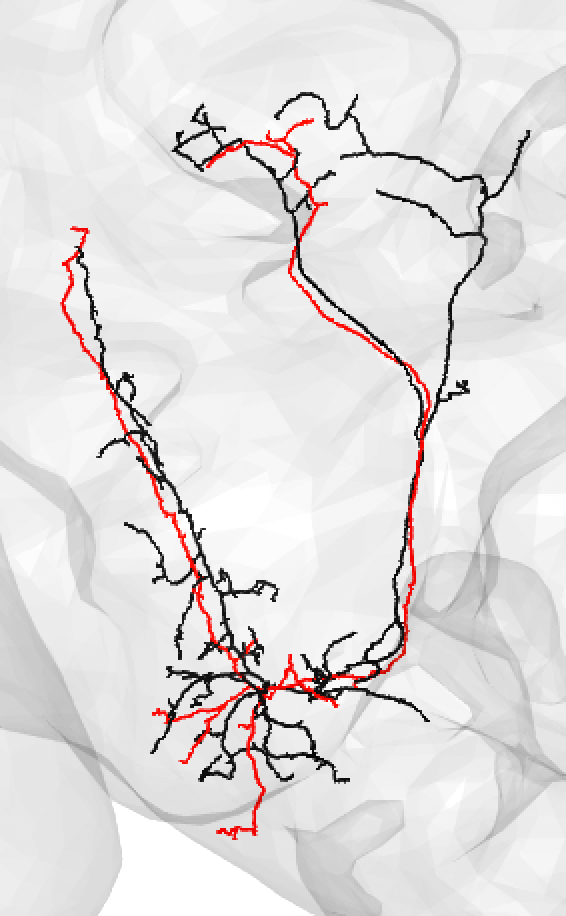 +
+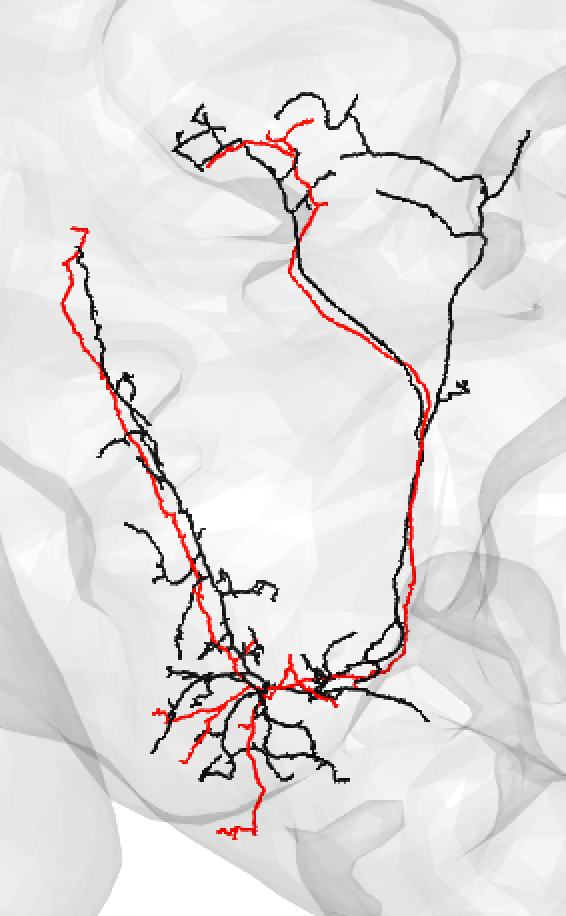
Or:
-  +
+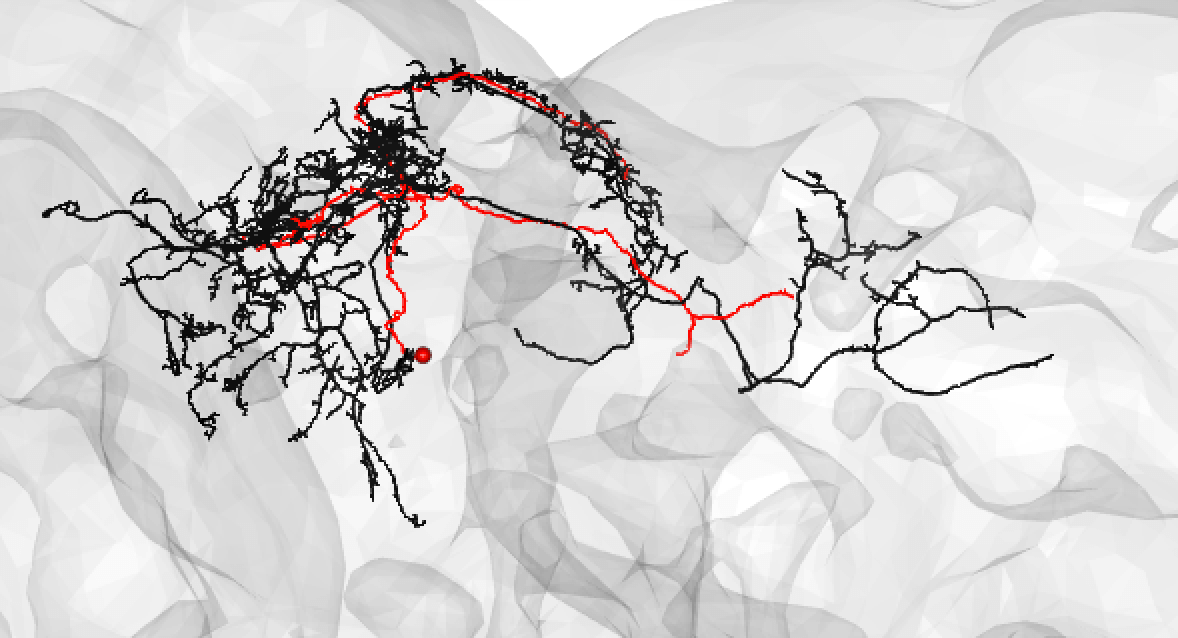
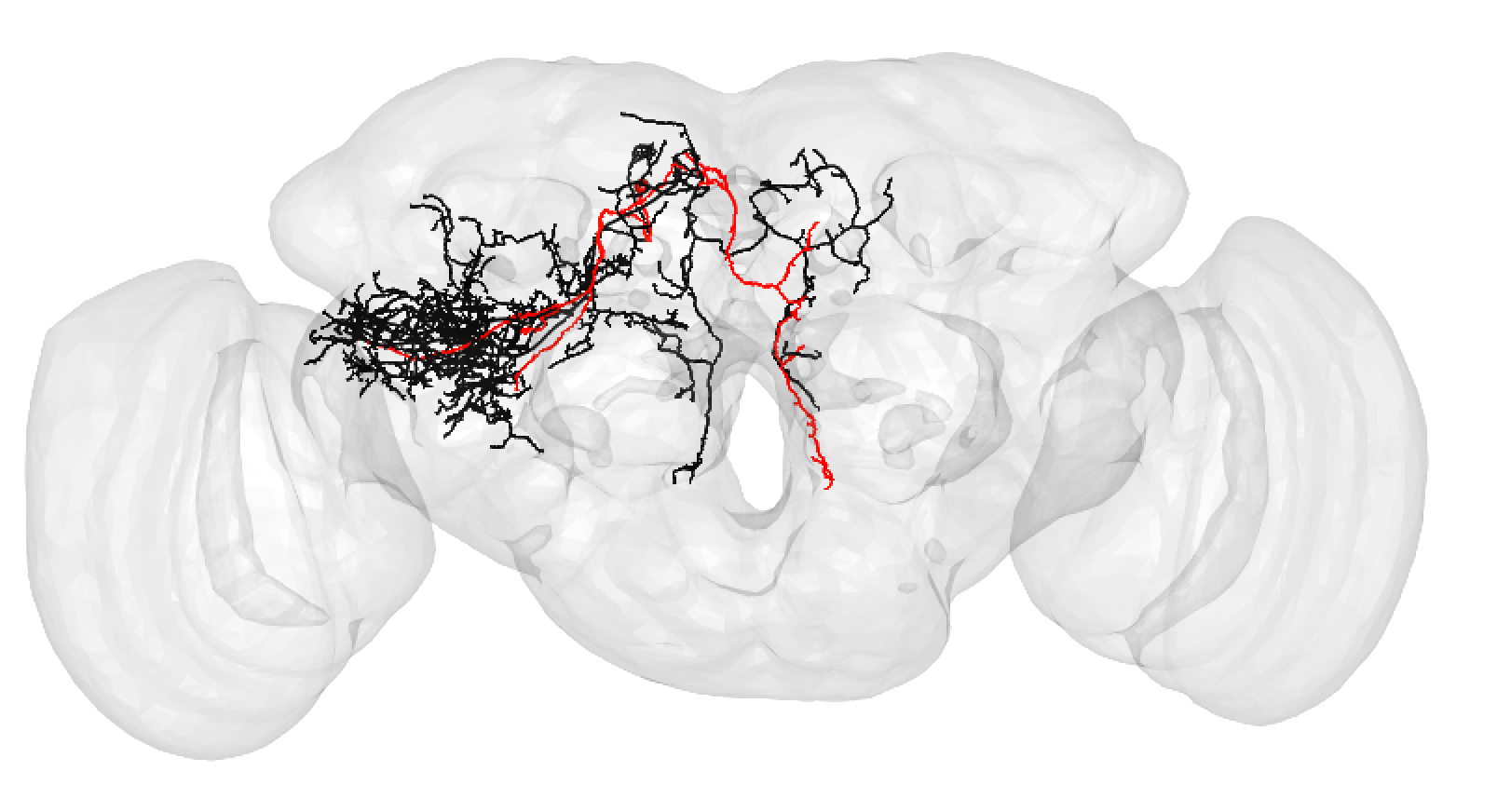
Bear in mind that the hemibrain volume only covers ~1/4 of the fly mid-brain, so neurons are truncated (here hemibrain neuron in black) but we can still make matches for many of them:
-  +
+
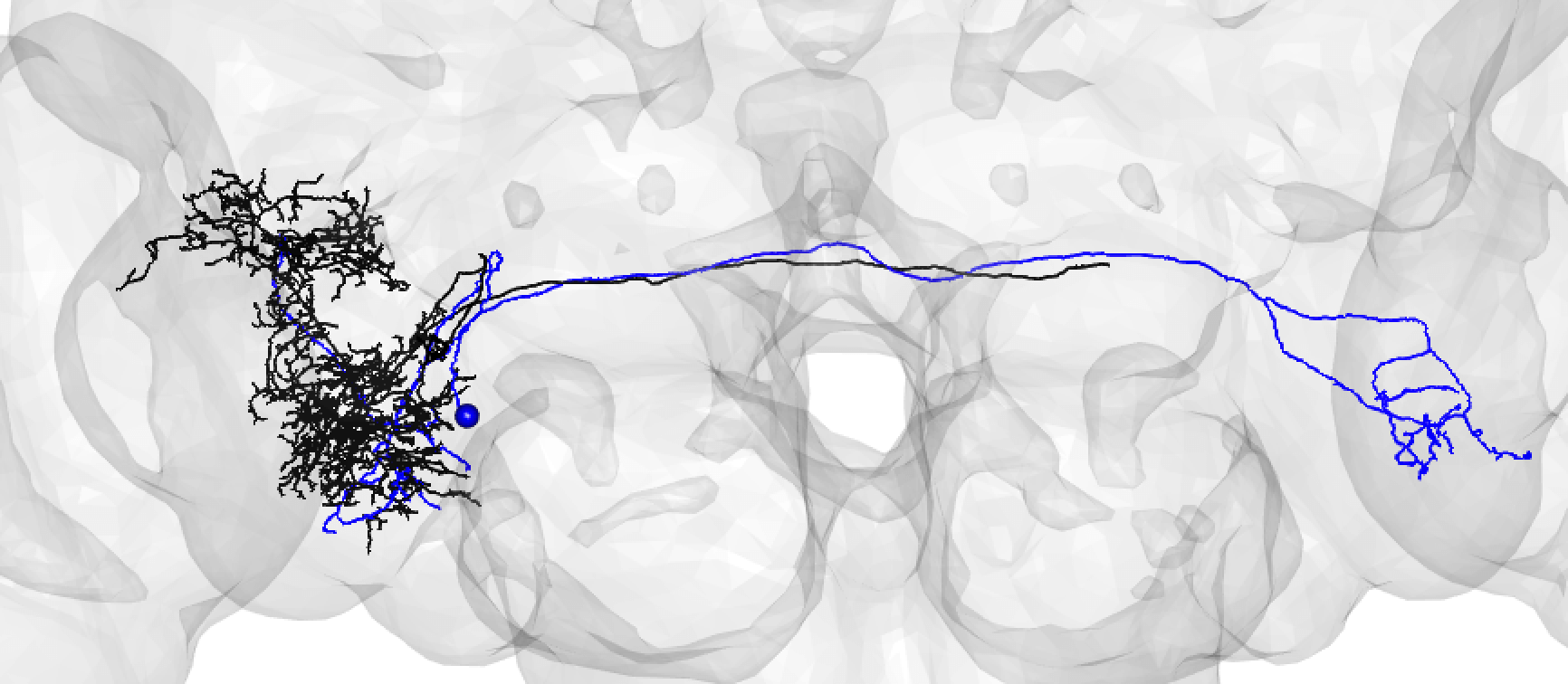
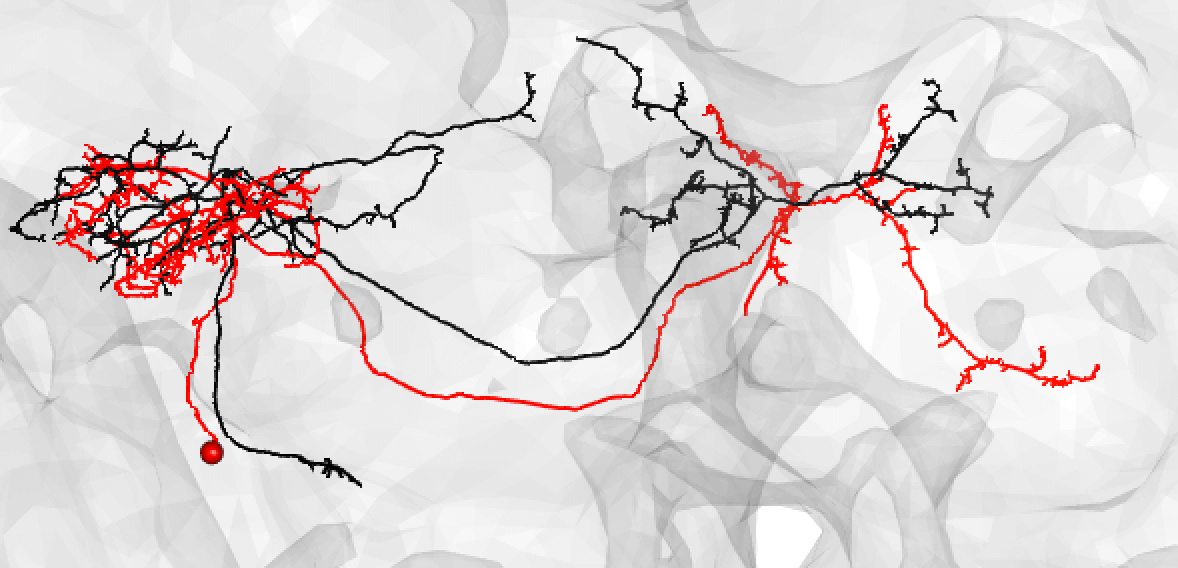
A larger degree of under-tracing may lead you to assign a match as poor. In this case, you think the two neurons may be ‘the same isomorphic cell type’ but you could be wrong. For example:
-  +
+
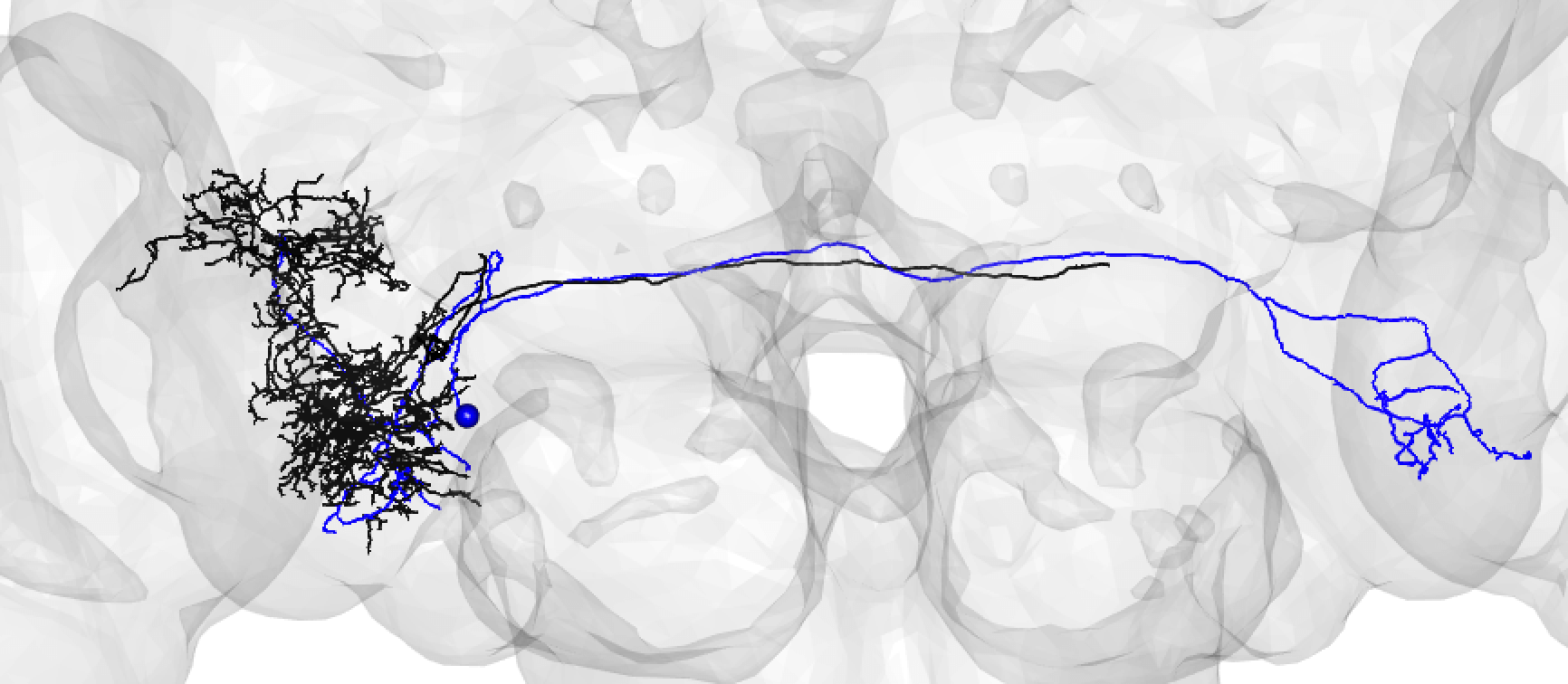
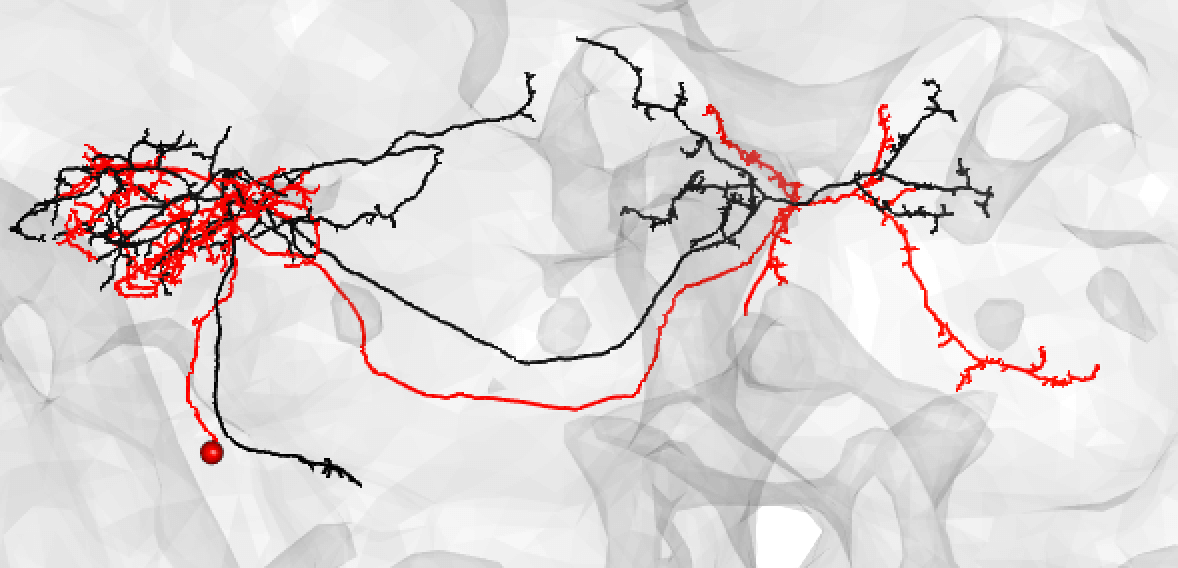
A poor match may also be made if you think there is a slight offset, possibly due to a registration issue:
-  +
+
Though in this case, choosing an even lesser-traced FAFB neuron may be better:
- 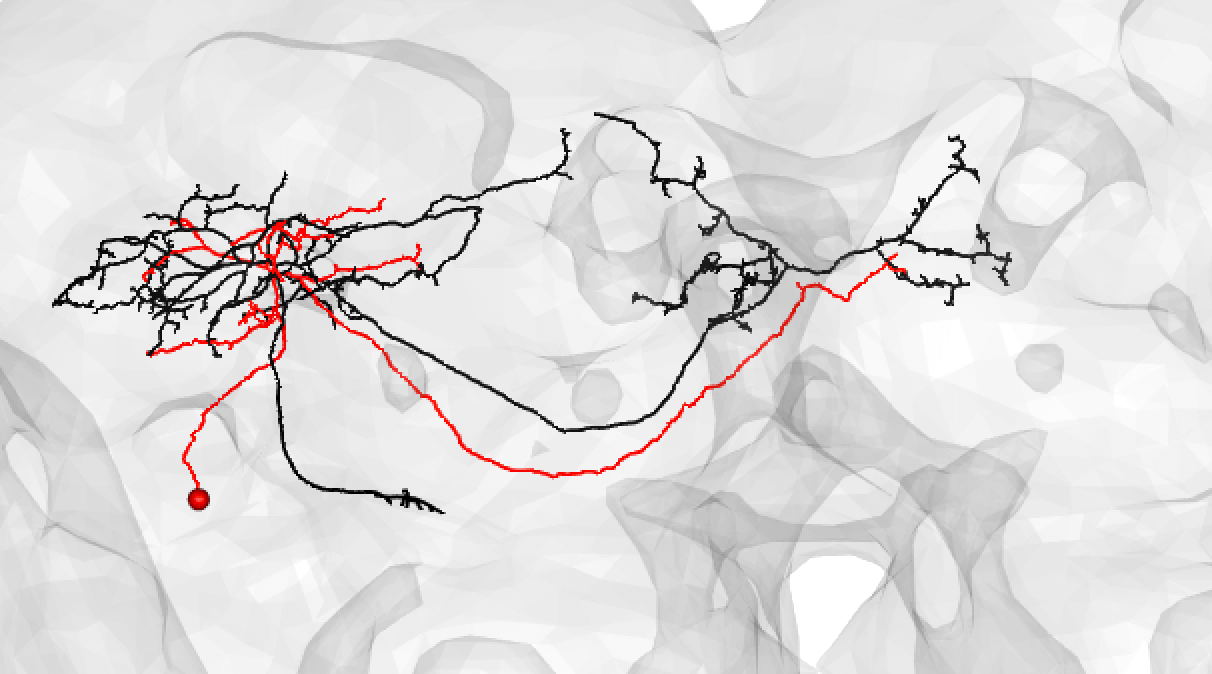 +
+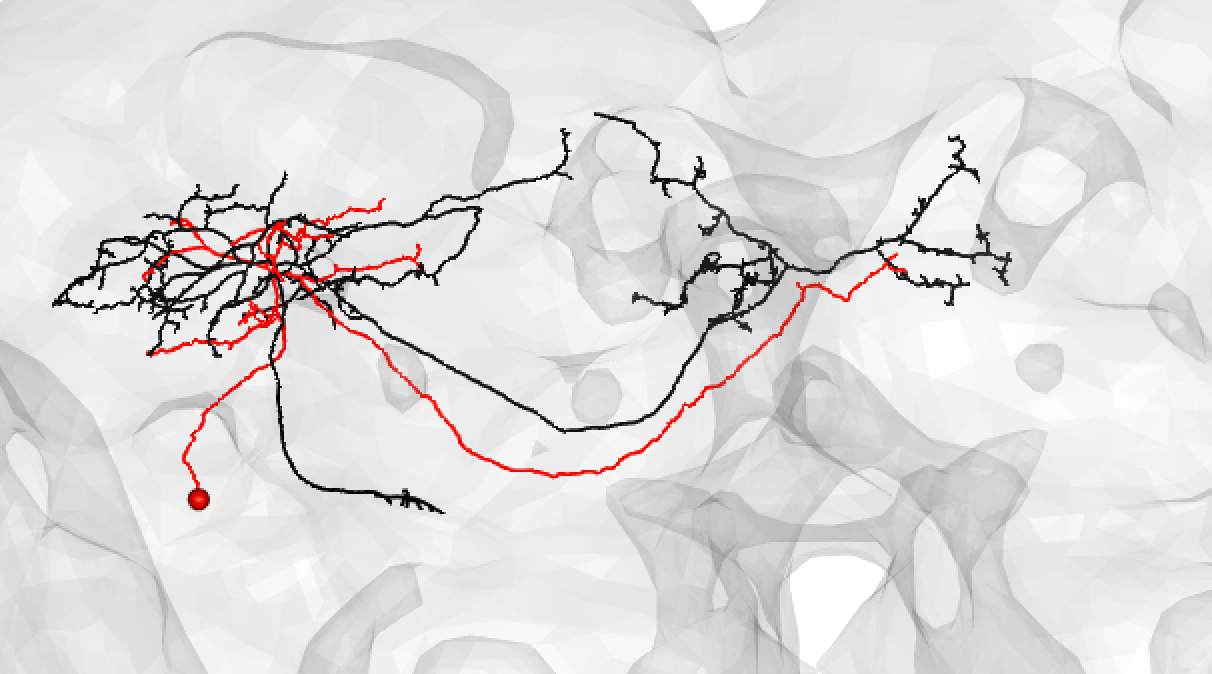
A poor match can be given even to very under-traced FAFB neurons:
- 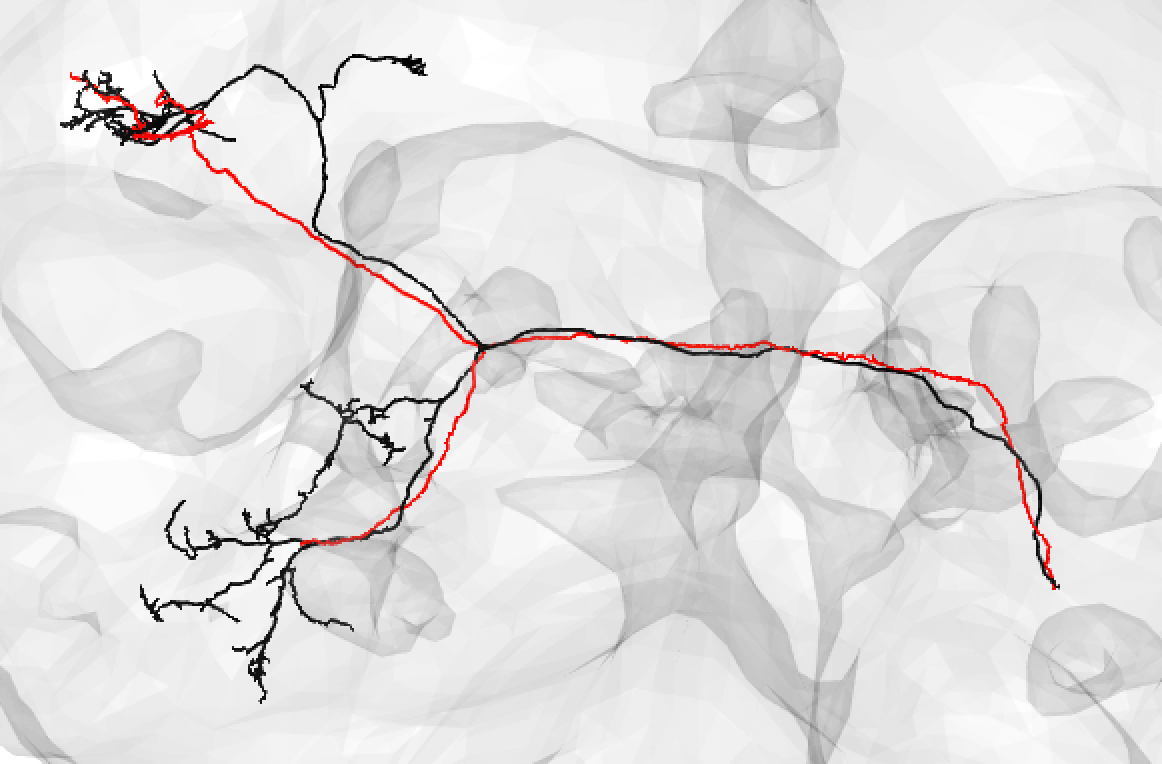 +
+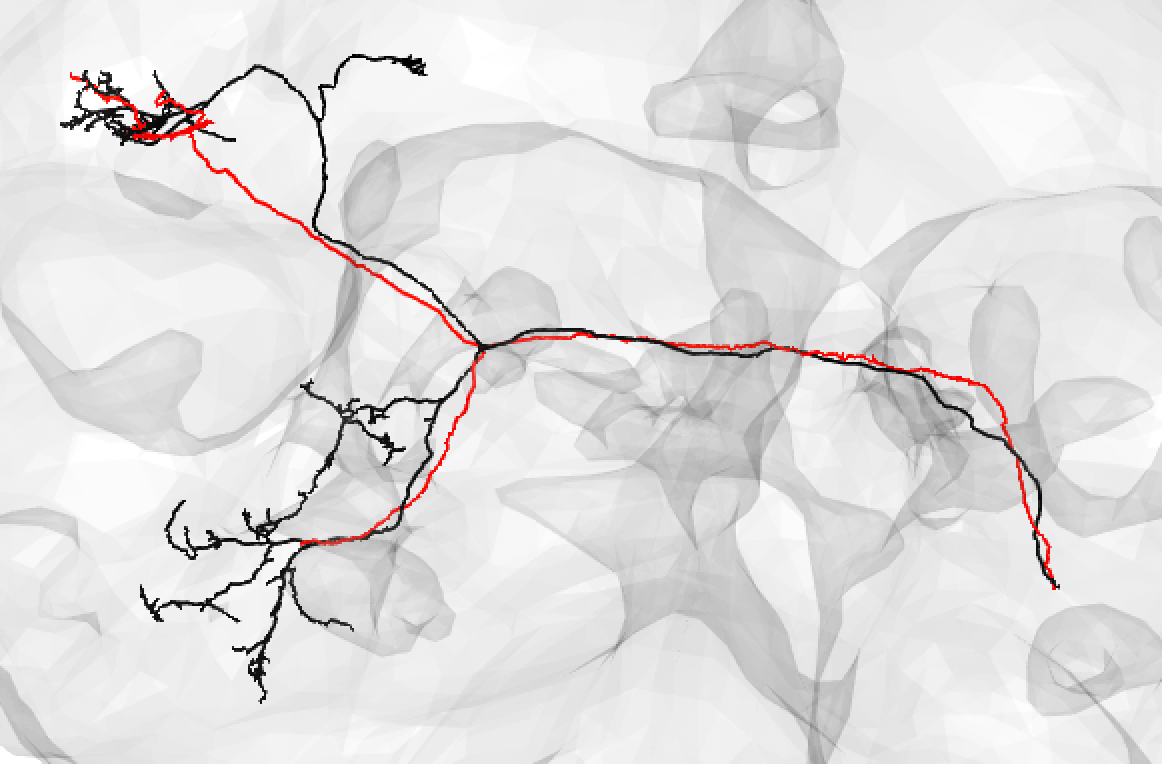
And even fragments if you are convinced the morphology is unique enough (but be careful!):
- 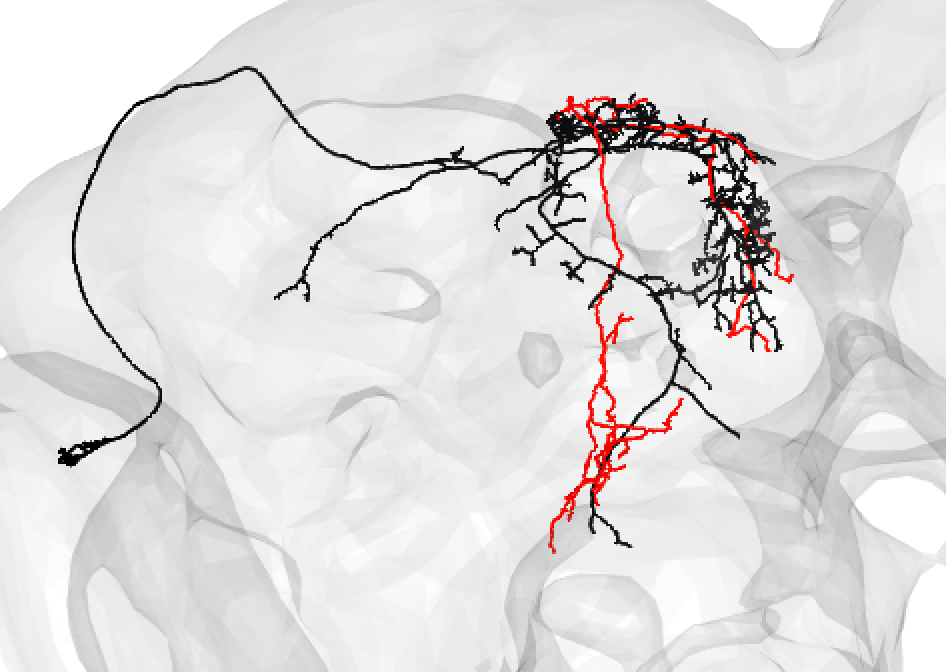 +
+
@@ -278,7 +281,7 @@
fafb_matching(ids = "16", overwrite = "none") # Re-look only if no proper match, or just a tract-only match, was found before.
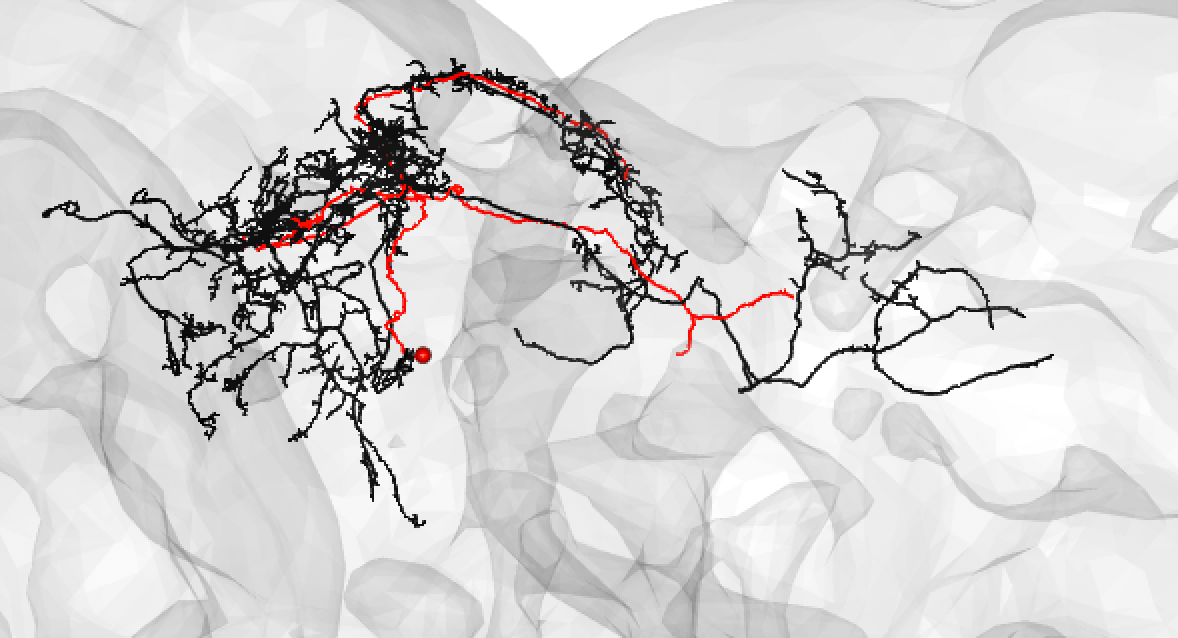
When you run these functions you will enter an interactive pipeline in an
rglwindow. Prompts will be given to you in your R console and you can rotate and pan in the window to see neurons. The neuron selected for-matching is shown in blue (i.e. if usinghemibrain_matchingthis will be a hemibrain neuron), and potential matches in red (i.e. if usinghemibrain_matchingthese will be FAFB neurons). Potential matches are shown by NBLAST score (a measure of morphological similarity). Usually, for reasonably traced FAFB neurons, a good match appears in the top 10 hits.-  +
+ @@ -306,7 +309,7 @@
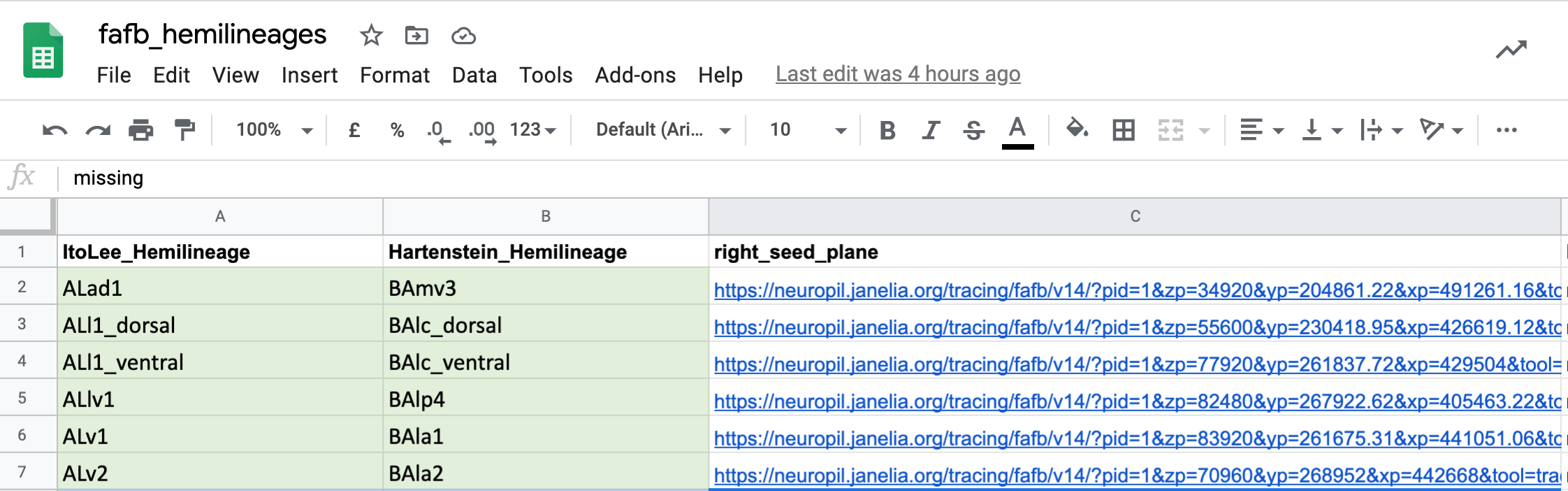
@@ -306,7 +309,7 @@Uses
One use we have already found for all of this match making, is to cross-identify neuron cell body fiber tracts and (hemi)lineages. This means that we now have the locations in FAFB for different known sets of cells. You can see seed planes for them here.
- 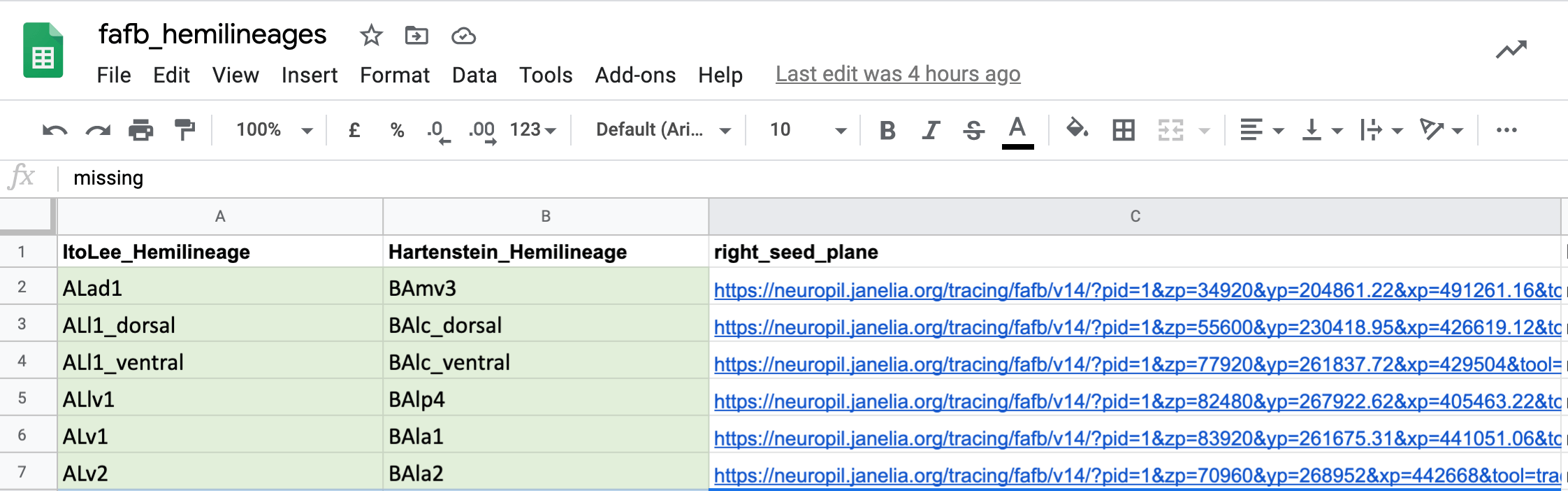 +
+
- + hemibrain and flywire meta data + +
- flywire
-
@@ -138,7 +141,7 @@
hemibrainr
-The goal of hemibrainr is to provide useful code for preprocessing and analysing data from the Janelia FlyEM hemibrain project. It contains specific functionality for splitting neurons into axons and dendrites, including split edgelists for connectivity. It also has the capability to load large amount of precomputed data on flywire, FAFB and hemibrain neurons from a linked Google drive. Please see this article.
+The goal of hemibrainr is to provide useful code for preprocessing and analysing data from the Janelia FlyEM hemibrain project. It contains specific functionality for splitting neurons into axons and dendrites, including split edgelists for connectivity. It also has the capability to load large amount of precomputed data on flywire, FAFB and hemibrain neurons from a linked Google drive. Please see this article.
@@ -303,8 +306,8 @@@@ -146,7 +149,7 @@
diff --git a/docs/authors.html b/docs/authors.html index 9f3a2e30..8ff2b467 100644 --- a/docs/authors.html +++ b/docs/authors.html @@ -95,6 +95,9 @@
-
+
- + hemibrain and flywire meta data +
- flywire diff --git a/docs/favicon-16x16.png b/docs/favicon-16x16.png index 99b47eff..410b11fa 100644 Binary files a/docs/favicon-16x16.png and b/docs/favicon-16x16.png differ diff --git a/docs/favicon-32x32.png b/docs/favicon-32x32.png index d4f44fea..84426d89 100644 Binary files a/docs/favicon-32x32.png and b/docs/favicon-32x32.png differ diff --git a/docs/index.html b/docs/index.html index eb1d4736..aef9c186 100644 --- a/docs/index.html +++ b/docs/index.html @@ -56,6 +56,9 @@
- + hemibrain and flywire meta data + +
- flywire
-
@@ -106,7 +109,7 @@
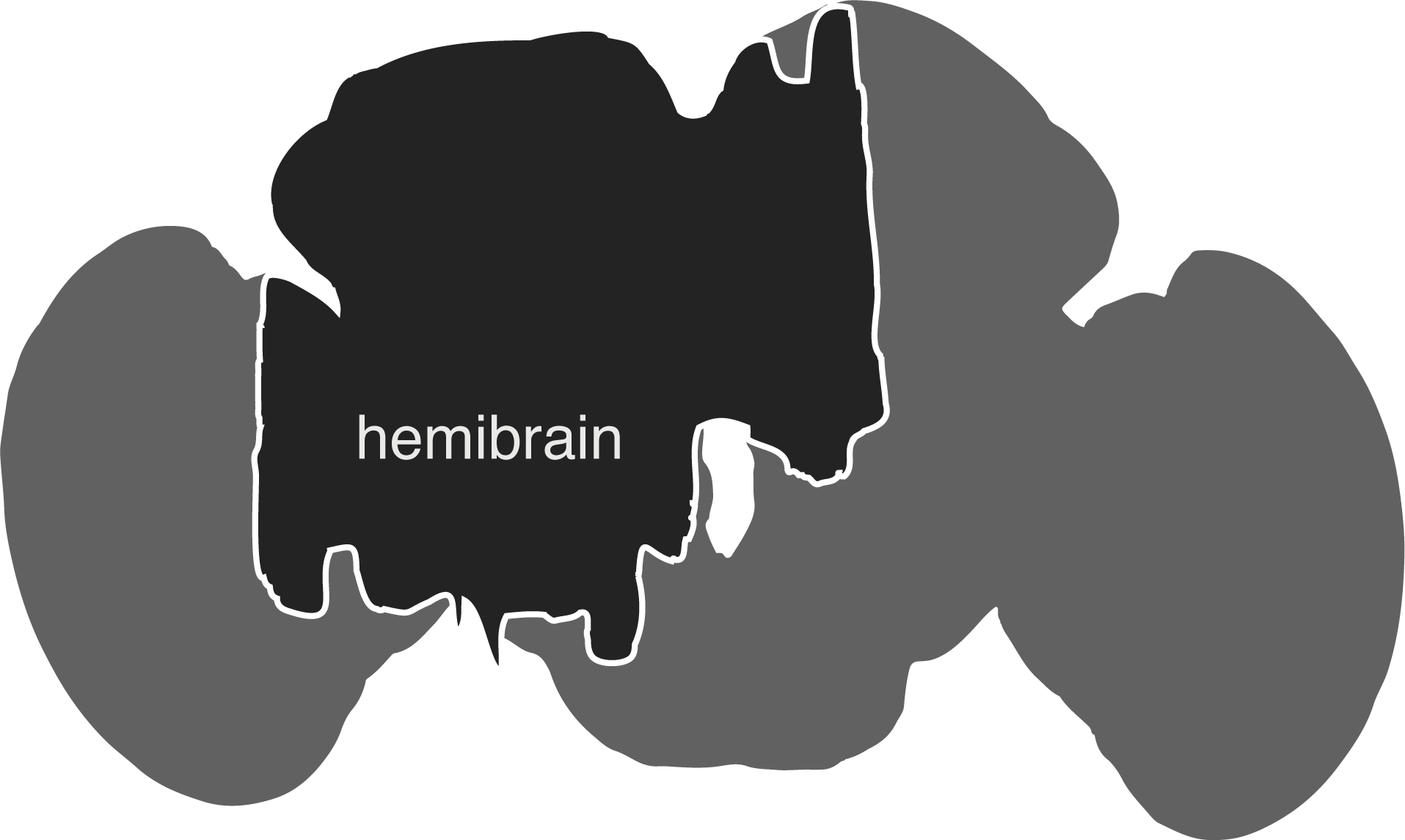
The goal of hemibrainr is to provide useful code for preprocessing and analysing data from the Janelia FlyEM hemibrain project. It makes use of the natverse R package, neuprintr to get hemibrain data from their connectome analysis and data hosting service neuprint. The dataset has been described here. Using this R package in concert with the natverse ecosystem is highly recommended.
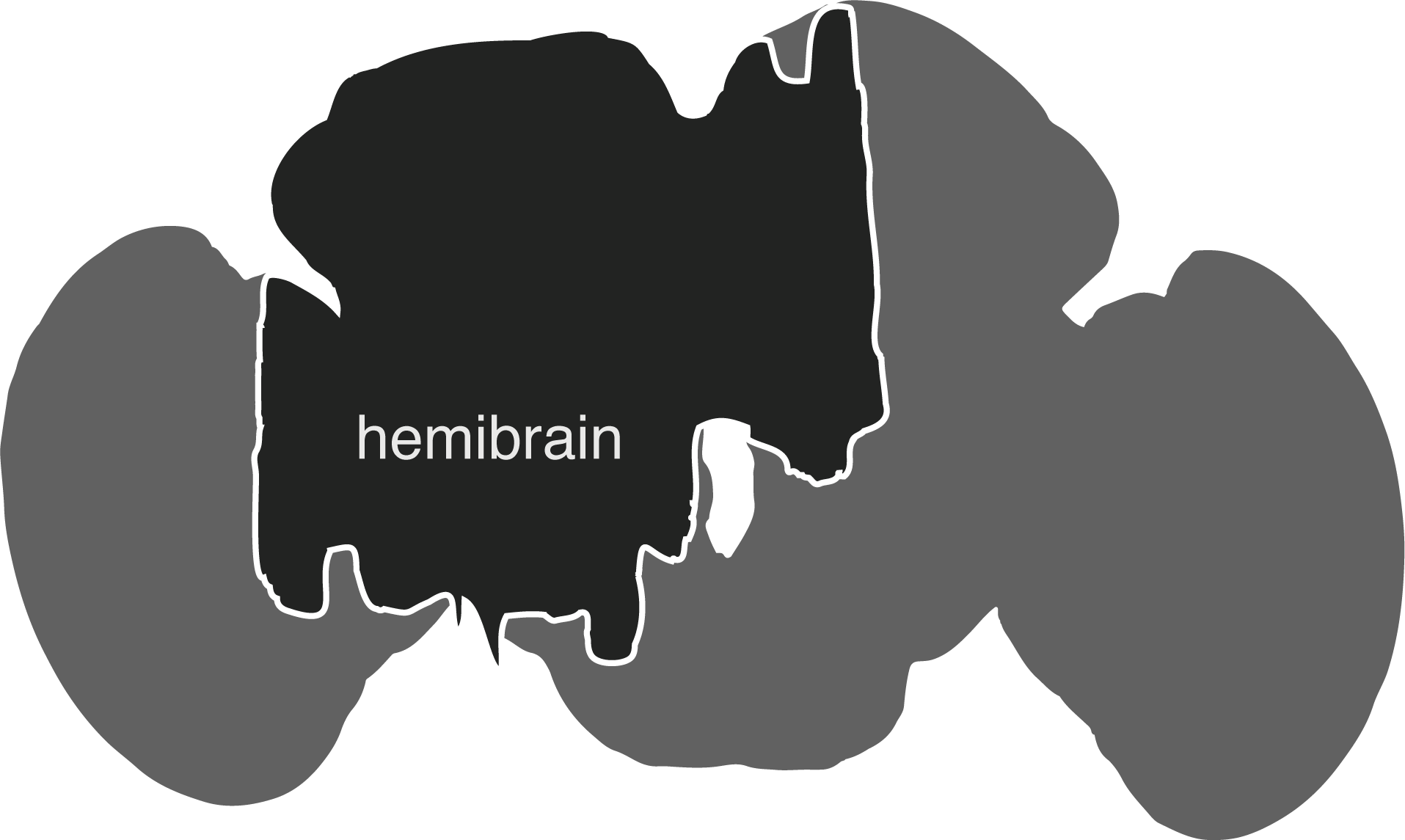
The hemibrain connectome comprises the region of the fly brain depicted below. It is ~21,662 ~full neurons, 9.5 million synapses and is about ~35% complete in this region:
-
+
 -
-@@ -117,7 +120,7 @@
@@ -184,7 +187,7 @@# install if (!require("remotes")) install.packages("remotes") -remotes::install_github("flyconnectome/hemibrainr") +remotes::install_github("natverse/hemibrainr") # use library(hemibrainr)For this, you need access to th hemibrainr google team drive. Authentication is through an email account. Once you have access, there are two basic ways to mount the data for use:
Option 1, mount your Google drives using Google filestream. However, for this to work you will need Google Workspace, Google’s monthly subscription offering for businesses and organizations. One the Google filestream application is run, you should be able to see your drives mounted like external hard drive, as so:
-
+
Then, this should work:
-@@ -194,7 +197,7 @@
# Now just get the name of your default team drive. ## This will be used to locate your team drive using the R package googledrive hemibrainr_team_drive()
Option 2, this is free. You still need authenticated access to the hemibrainr Gogle team drive. It cna then be moutned using rclone. First, download rclone for your operating system. You can also download from your system’s command line (e.g. from terminal) and then configure it for the drive:
+Option 2, this is free. You still need authenticated access to the hemibrainr Gogle team drive. It can then be mounted using rclone. First, download rclone for your operating system. You can also download from your system’s command line (e.g. from terminal) and then configure it for the drive:
@@ -216,7 +219,7 @@# And now we are back to: options("Gdrive_hemibrain_data")
For more detailed instructions, see this article.
+For more detailed instructions, see this article.
@@ -270,7 +273,7 @@
This package was created by Alexander Shakeel Bates and Gregory Jefferis. You can cite this package as:
-citation(package = "hemibrainr")Bates AS, Jefferis GSXE (2020). hemibrainr: Code for working with data from Janelia FlyEM’s hemibrain project. R package version 0.1.0. https://github.com/flyconnectome/hemibrainr
+Bates AS, Jefferis GSXE (2020). hemibrainr: Code for working with data from Janelia FlyEM’s hemibrain project. R package version 0.1.0. https://github.com/natverse/hemibrainr
Developers
diff --git a/docs/pkgdown.yml b/docs/pkgdown.yml index ca80c1d6..d7b4900f 100644 --- a/docs/pkgdown.yml +++ b/docs/pkgdown.yml @@ -2,6 +2,7 @@ pandoc: 2.3.1 pkgdown: 1.6.1.9000 pkgdown_sha: 6094ac3dd80a1f3f1d4e2f47d0ed4716d51362b0 articles: + data: data.html flywire: flywire.html google_filestream: google_filestream.html hemibrain_alpns_toons: hemibrain_alpns_toons.html @@ -9,5 +10,5 @@ articles: hemibrain_connectivity: hemibrain_connectivity.html match_making: match_making.html packages_guide: packages_guide.html -last_built: 2020-12-04T19:45Z +last_built: 2021-01-06T17:54Z diff --git a/docs/reference/add_Label.html b/docs/reference/add_Label.html index 7b210334..e83b2195 100644 --- a/docs/reference/add_Label.html +++ b/docs/reference/add_Label.html @@ -97,6 +97,9 @@Dev status
-
-
 -
- +
+ +
+

All vignettes
-
+
FormatAn object of class
characterof length 307.An object of class
characterof length 2383.An object of class
-characterof length 1428.An object of class
+characterof length 431.An object of class
characterof length 345.An object of class
characterof length 1927.An object of class
characterof length 5.An object of class
characterof length 24.An object of class
-characterof length 2129.An object of class
characterof length 1927.An object of class
data.framewith 2651 rows and 44 columns.Details
diff --git a/docs/reference/extract_cable.html b/docs/reference/extract_cable.html index b122bb0e..d1721dbc 100644 --- a/docs/reference/extract_cable.html +++ b/docs/reference/extract_cable.html @@ -96,6 +96,9 @@fafb_hemibrain_annotate(x, flywire = TRUE, ...) diff --git a/docs/reference/flow_centrality.html b/docs/reference/flow_centrality.html index d31da248..b0cfd07d 100644 --- a/docs/reference/flow_centrality.html +++ b/docs/reference/flow_centrality.html @@ -100,6 +100,9 @@
#> Error in neuprintr::neuprint_read_neurons(tough): Error reading bodyids# Now make sure the neurons have a soma marked ## Some hemibrain neurons do not, as the soma was chopped off neurons.checked = hemibrain_skeleton_check(neurons, meshes = hemibrain.surf) -#>#>#> Warning: invalid factor level, NA generated#> Warning: invalid factor level, NA generated#> Warning: invalid factor level, NA generated#> Warning: invalid factor level, NA generated#> Warning: invalid factor level, NA generated#> Warning: invalid factor level, NA generated+#> Error in nat::as.neuronlist(x): object 'neurons' not found# Split neuron ## These are the recommended parameters for hemibrain neurons neurons.flow = flow_centrality(neurons.checked, polypre = TRUE, mode = "centrifugal", split = "distance") - +#> Error in flow_centrality(neurons.checked, polypre = TRUE, mode = "centrifugal", split = "distance"): object 'neurons.checked' not foundif (FALSE) { # Plot the split to check it nat::nopen3d() diff --git a/docs/reference/flycircuit_neurons.html b/docs/reference/flycircuit_neurons.html index 08781a2e..c1505f02 100644 --- a/docs/reference/flycircuit_neurons.html +++ b/docs/reference/flycircuit_neurons.html @@ -98,6 +98,9 @@-flywire_meta(local = FALSE, folder = "flywire_neurons/", sql = TRUE, ...) +
flywire_meta(local = FALSE, folder = "flywire_neurons/", sql = FALSE, ...) + +flywire_failed(local = FALSE, folder = "flywire_neurons/", sql = FALSE, ...) flywire_contributions( local = FALSE, @@ -168,7 +173,7 @@
+flywire_ids(local = FALSE, folder = "flywire_neurons/", sql = FALSE, ...)Read precomputed flywire data from the hemibrainr Google Drive
... ) -flywire_ids(local = FALSE, folder = "flywire_neurons/", sql = TRUE, ...)Arguments
diff --git a/docs/reference/flywire_ids_update.html b/docs/reference/flywire_ids_update.html index c2aa7e04..212d3b4d 100644 --- a/docs/reference/flywire_ids_update.html +++ b/docs/reference/flywire_ids_update.html @@ -47,7 +47,7 @@ - @@ -99,6 +99,9 @@
diff --git a/docs/reference/paper_colours.html b/docs/reference/paper_colours.html index cb9380ac..e0818b69 100644 --- a/docs/reference/paper_colours.html +++ b/docs/reference/paper_colours.html @@ -96,6 +96,9 @@