For System Administrators: Installation
- OS: Linux
- Environment: Perl (>=5.10), PHP (>=5.3) together with HTTP service
- Primer3 (>=2.3).
Please set up Primer3 carefully to make sure it works properly, especially the PRIMER_THERMODYNAMIC_PARAMETERS_PATH
- Samtools (>=1.2).
- NCBI BLAST+ (>=2.2.18)
It must be BLAST+, not the legacy BLAST
- Clone the repository to a location where your HTTP server can visit
$ cd /var/www/html/
$ git clone https://github.com/billzt/PrimerServer.git-
(Important) Make the directory
savewritable by your HTTP server. For example, if you are using Apache HTTP server on CentOS system, you might try:
$ cd PrimerServer
$ sudo chgrp apache save
$ sudo chmod 775 saveFor other HTTP server (such as Nigix) or other Linux distribution (such as Ubuntu), please refer to their documents
- Copy the configuration file as
config.iniand do some necessary configuration.
$ cp config.sample.ini config.ini- Visit on your web browser as: http://your.domain/PrimerServer
- Software Path (Absolute):
samtools = "/path/to/samtools"
primer3 = "/path/to/primer3_core"
blastn = "/path/to/blastn"
makeblastdb = "/path/to/makeblastdb"- NCBI BLAST Database Directory (Absolute):
database = "/path/to/db"For tests, you can copy the example files in directory
test_datato/path/to/db. In order to learn how to build your own BLAST database, see below.
- System Configuration
useCPU = 20The number of CPUs used by PrimerServer. Suggesting to set it as many as possible, as this would strongly affect the running time.
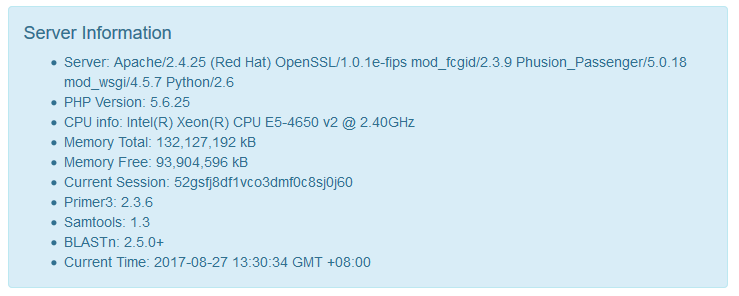
showInfo = falseWhether to show detailed information of your server on the web UI. If you are deploying a product app, please turn it to false.

If you are deploying a developing or intranet app, you can turn it to true.
removeTmp = trueWhether to delete the tempory files after users closing the window. If you want to debug, please turn it to false.
limitSite = 100The maximum number of input sites in a single run when design primers. Lower it if your server is poor.
limitPrimer = 1000The maximum number of input primers in a single run when directly checking specificity. Lower it if your server is poor.
limitDatabase = 4The maximum number of selected databases when doing specificity checking. Lower it if your server is poor.
; Database Alias
[Database.Cateogry1]
File1 = "Alias1"
File2 = "Alias2"
[Database.Cateogry2]
File3 = "Alias3"
File4 = "Alias4"
[Database.Example]
example.fa = "Example Database"All the database items would be shown in a selection list. If you want to group your databases (For example, group by animal, plant, fungi, ...), you can add this setting. The keys should be database names and must be simple words (avoiding any special characters), and the values can be any descriptive words.
A test database (including two sequences) has already been placed in the test_data directory. Copy all the five files to your database directory specified in the file config.ini
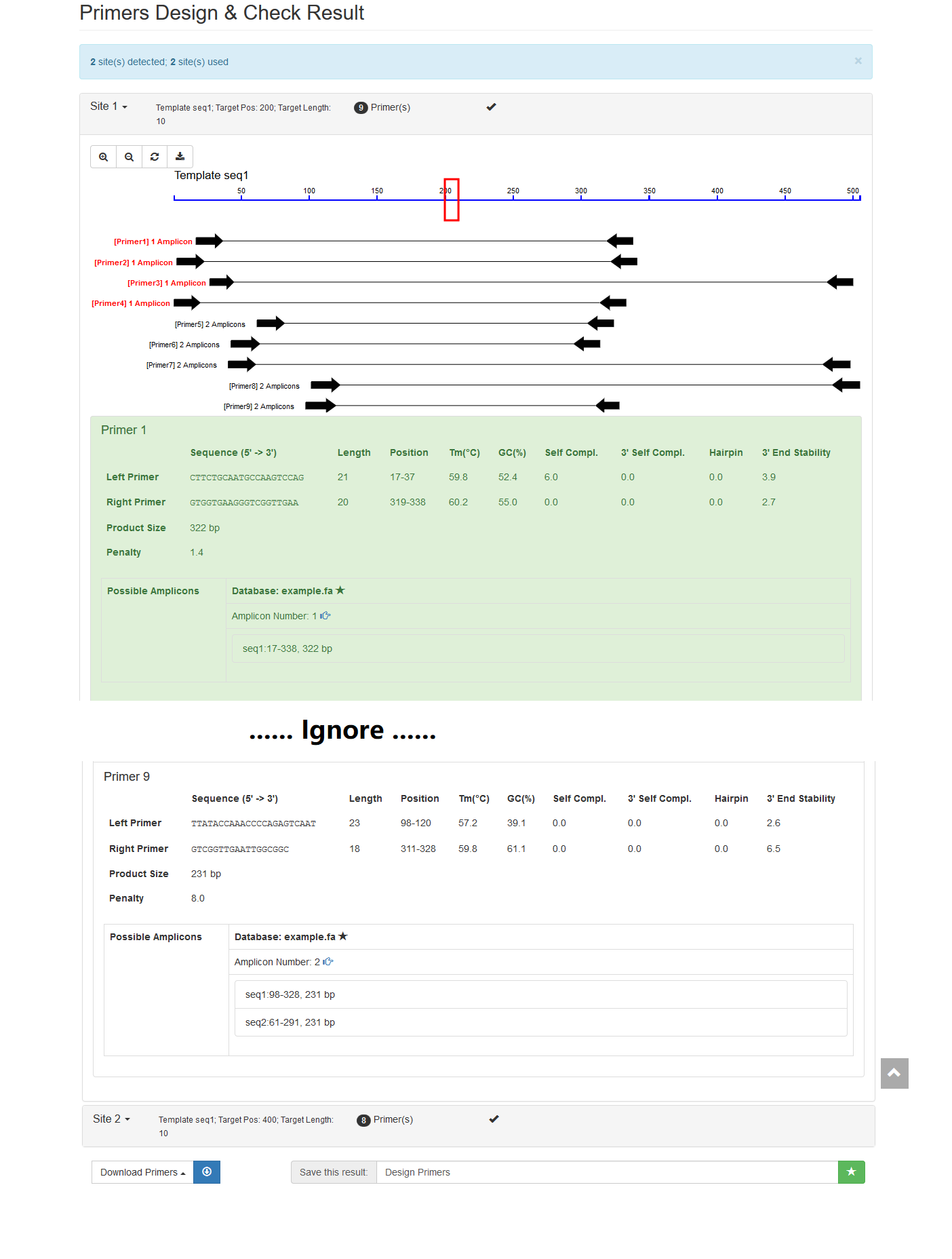
- Select the template database: Example Database
- Input Sites:
seq1 200 10
seq1 400 10- Select the checking database: Example Database
- Run (time costs: ~2s)
If you see results like this, then it is OK
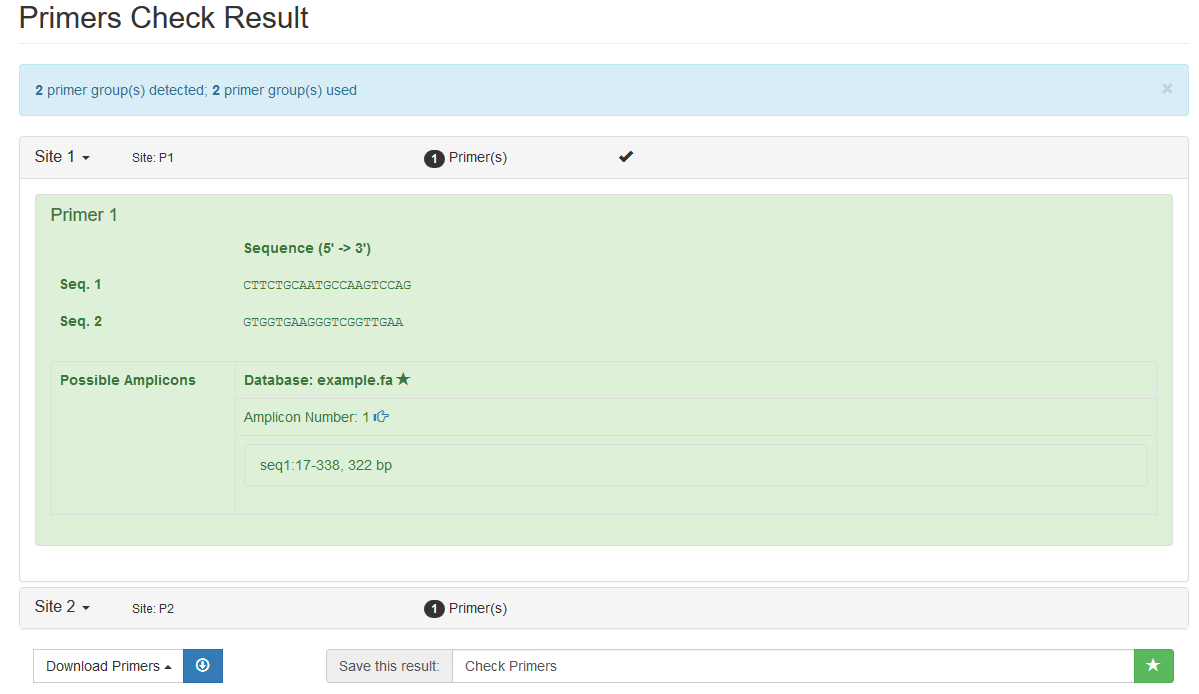
- Input Primer sequences:
P1 CTTCTGCAATGCCAAGTCCAG GTGGTGAAGGGTCGGTTGAA
P2 ACCAAACCCCAGAGTCAATTAA TCTATCTATTGCACTGCCTGTTG- Select the checking database: Example Database
- Run (time costs: ~0.5s)
If you see results like this, then it is OK
After running, click the save icon in the bottom. A modal would pop out to confirm your results. After confirmation, you would be taken to the result's page. If you see a table like this, then it is OK
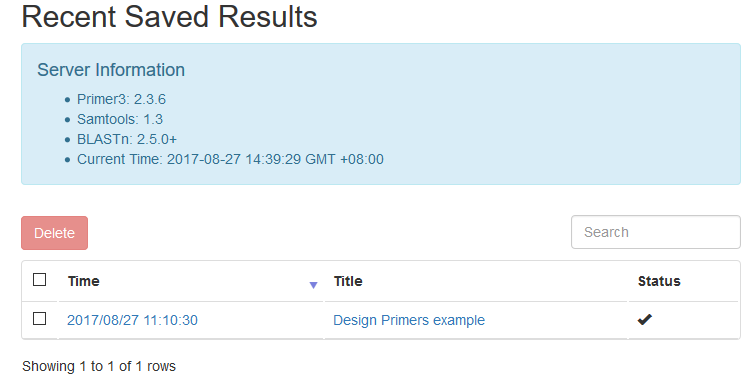
To prepare new databases, you need a FASTA file containing the genome (or cDNA) sequences. Place it in your database directory specified in the file config.ini. Then:
$ samtools faidx genome.fa
$ makeblastdb -dbtype nucl -in genome.faAfter this finished, you can add alias to your file config.ini.
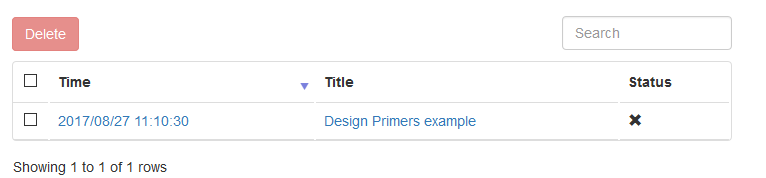
Each result saved by users would be created as a HTML file in the save directory. As a System Administrator, you can delete files such as those too old (for example, ten years ago, 😄). Files deleted by system administrators would represented as a ❎ mark such as: